生信人 RNA-seq
RNA-seq:6-qc-2
#先跑一个综合的 Multiqc 报告
#将fastqc生成的多个报告整合成一个报告,方便查看所有测序数据的质量。
conda install multiqc -c https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
cd /home/yinwen/RNA-seq_report/
multiqc .
![]()
用我们自己的DG数据分析一下:
① General Statistics:
?在这里我们能看到各个样本的概况或基本信息

解读:
- %Dups:Duplicate Reads Percent,重复reads的比例
- %GC:Average %GC Content,平均GC含量百分比
- M Seqs:Total Sequences,总测序量
- Length:Average Sequence Length,平均序列长度
- %Failed:Percentage of modules failed in FastQC report,报告中不合格数据的百分比
点击左侧的?Configure Columns?可以自定义展示列参数

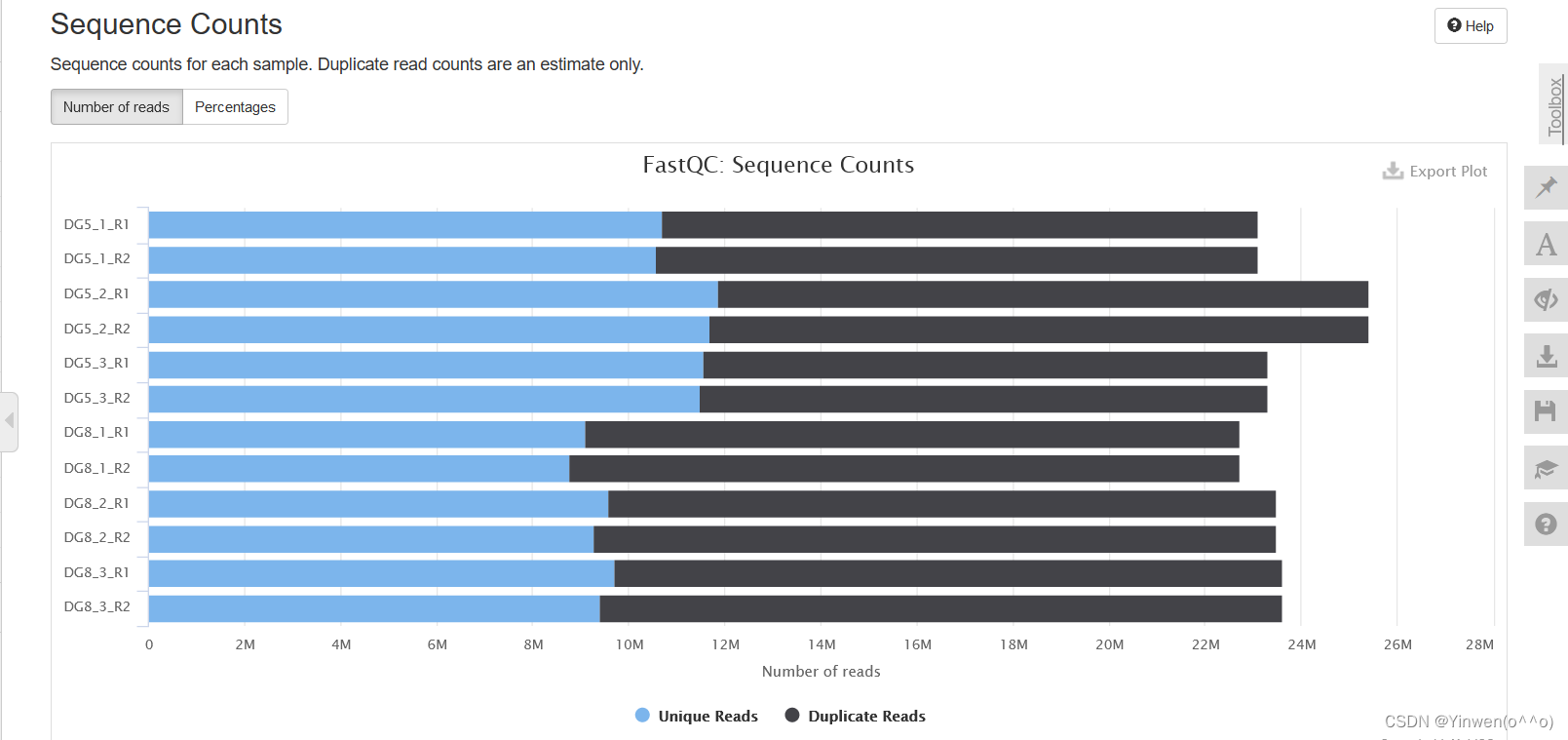
② Sequence Counts
该部分对每个样本序列进行了计数,横坐标为总的reads数(和General Stats中的M seqs一致),纵坐标为不同样本。蓝色和黑色下方有标记。
③?Sequence Quality Histograms

此部分为reads中每个位置(从0到150bp)的平均质量值,横坐标为位置。X轴并不是均匀的;纵坐标为质量分数,计算公式为

所以当质量分数为40的时候,p就是0.0001。
图中绿色表示合格(通过),黄色代表(警告),红色则代表失败(不合格)。
图上所示DG的综合数据的质量分数在35%~40%之间,说明p值低,错误率低,质量好。
④?Per Sequence Quality Scores
该部分为reads次数和平均质量分数之间的关系,可以理解为reads质量的分布情况,当质量小于27时报“警告”,小于20时报“失败”。

由图中可以看出来:
峰值越靠右代表高质量的reads越多,数据也就越好,说明我们的DG数据质量好
⑤ Per Base Sequence Content


MultiQC:能很直观的看到哪些样本是“WARN”,哪些是“FAIL”。当把鼠标放到图上时,还能清楚的看到每一个位置碱基的分布情况。
FastQC:该部分展现了reads每一个位置的ATCG四种碱基的分布情况。
· 横轴为位置,纵轴为碱基含量,正常情况下每个位置每种碱基出现的概率是相近的,四条线应该平行且相近。当部分位置碱基的比例出现分歧时,即四条线在某些位置纷乱交织,往往提示我们有污染。当任一位置的A/T比例与G/C比例相差超过10%,报"WARN";当任一位置的A/T比例与G/C比例相差超过20%,报"FAIL"。
分析:我们的DG的图前15bp,碱基频率有明显的差别,按道理说明有污染。但是,在大多数RNAseq文库制备方法中,前10-15bp碱基分布明显不均匀,这是正常的,具体取决于使用的文库试剂盒的类型。即使序列完全正确,这种碱基组成不均匀的数据也会被认为是不合格。
⑥ Per Sequence GC Content(用于评估污染)
该部分展现了reads的平均GC含量,我们能看到有12个“警告”。
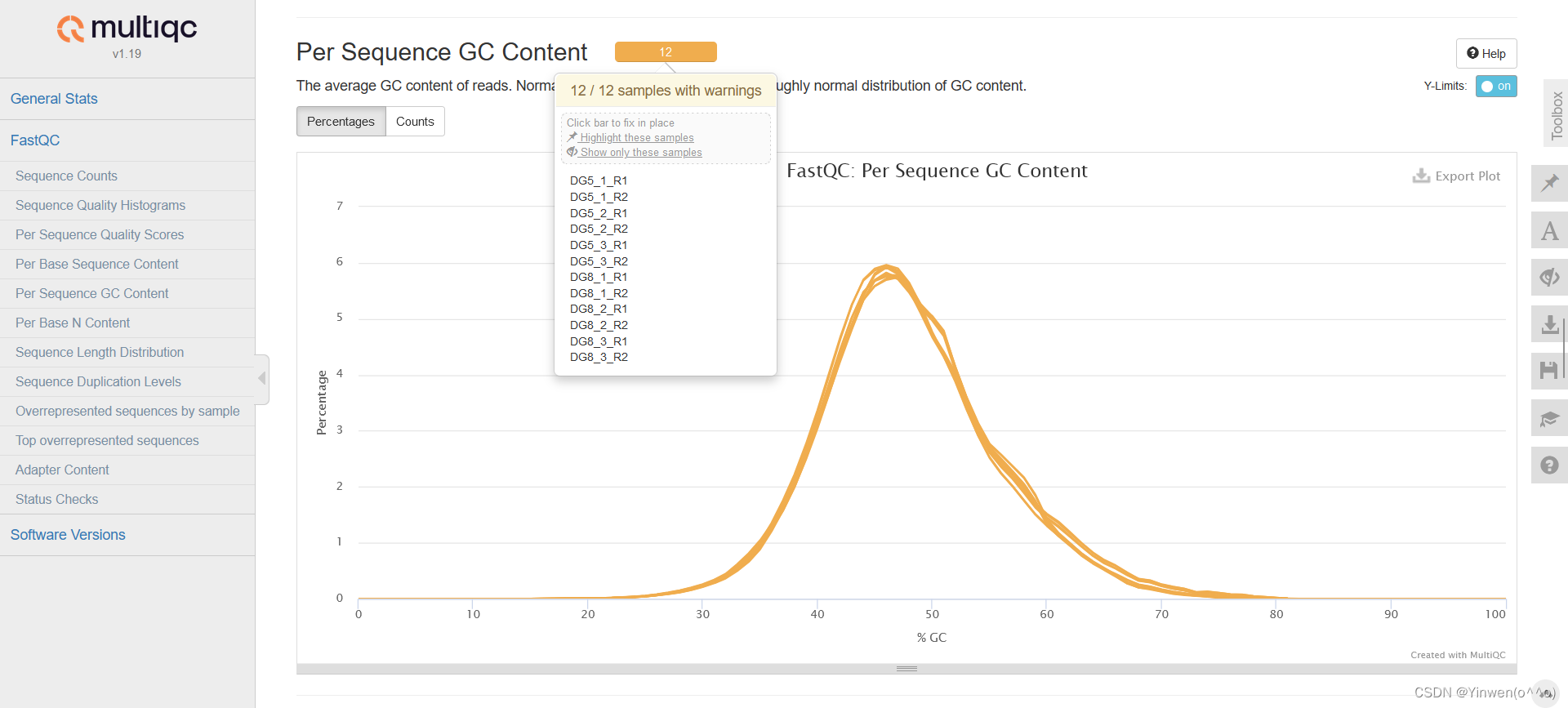

上图的fastqc报告来自我们非常高质量的RNA-seq数据,但FastQC仍然认定为“警告”,因为它比理论曲线窄。这种情况非常正常,因此可以忽略警告。
⑦?Per Base N Content
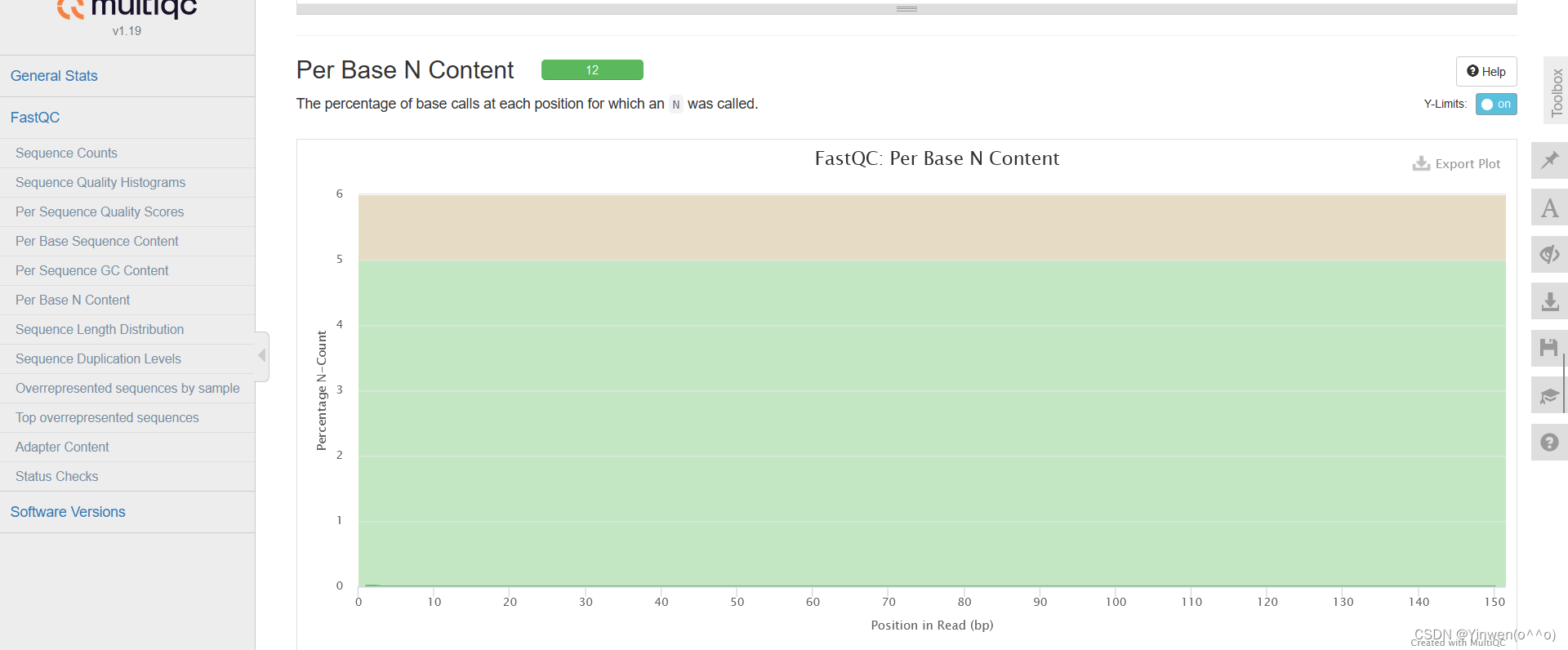
该部分展现了不同样本不同位置N的比例。当测序仪器不能辨别某条reads的某个位置到底是什么碱基时,就会产生“N”正常情况下,N值非常小。当任意位置的N的比例超过5%,报"WARN";当任意位置的N的比例超过20%,报"FAIL"。
⑧?Sequence Length Distribution

该部分为reads的长度分布,当reads长度不一致时报"WARN";当有长度为0的reads时报“FAIL”。
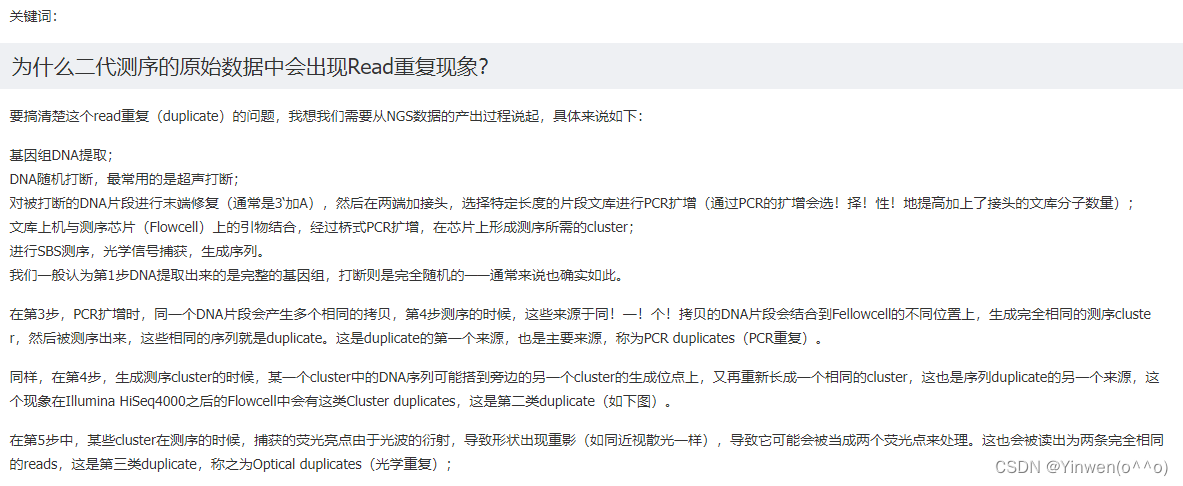
⑨?Sequence Duplication Levels

MultiQC报告的结果 12个都为FAIL
该部分展现了不同拷贝数的reads的频率。横坐标是duplication的次数,纵坐标是Deduplicated reads的百分比,以unique reads的总数作为100%。
当非unique的reads占总数的比例大于20%时,报"WARN";
当非unique的reads占总数的比例大于50%时,报"FAIL“。
扩展:通常有两种重复reads的来源,PCR重复即 biased PCR富集 和 真正高表达的序列。前者会错误的反映样本序列中的真实比例,后者是正常情况。测序深度越高,越容易产生一定程度的duplication;但如果duplication的程度很高,就提示我们可能有bias的存在。
接下来是过滤
①下载trim_galore
?#查找自己已经建立的环境
conda info --envs
#进入python2的环境
conda activate py2env
#查看依赖的安装包是否下载好
cutadapt -h
fastqc -h
#下载trim-galore
conda install trim-galore
# 查看是否安装好
trim_galore --help

#我们回到报告中查看接头,截取一段
zcat DG5_1_R1.fq.gz |grep TTCTAAGTTCATCT

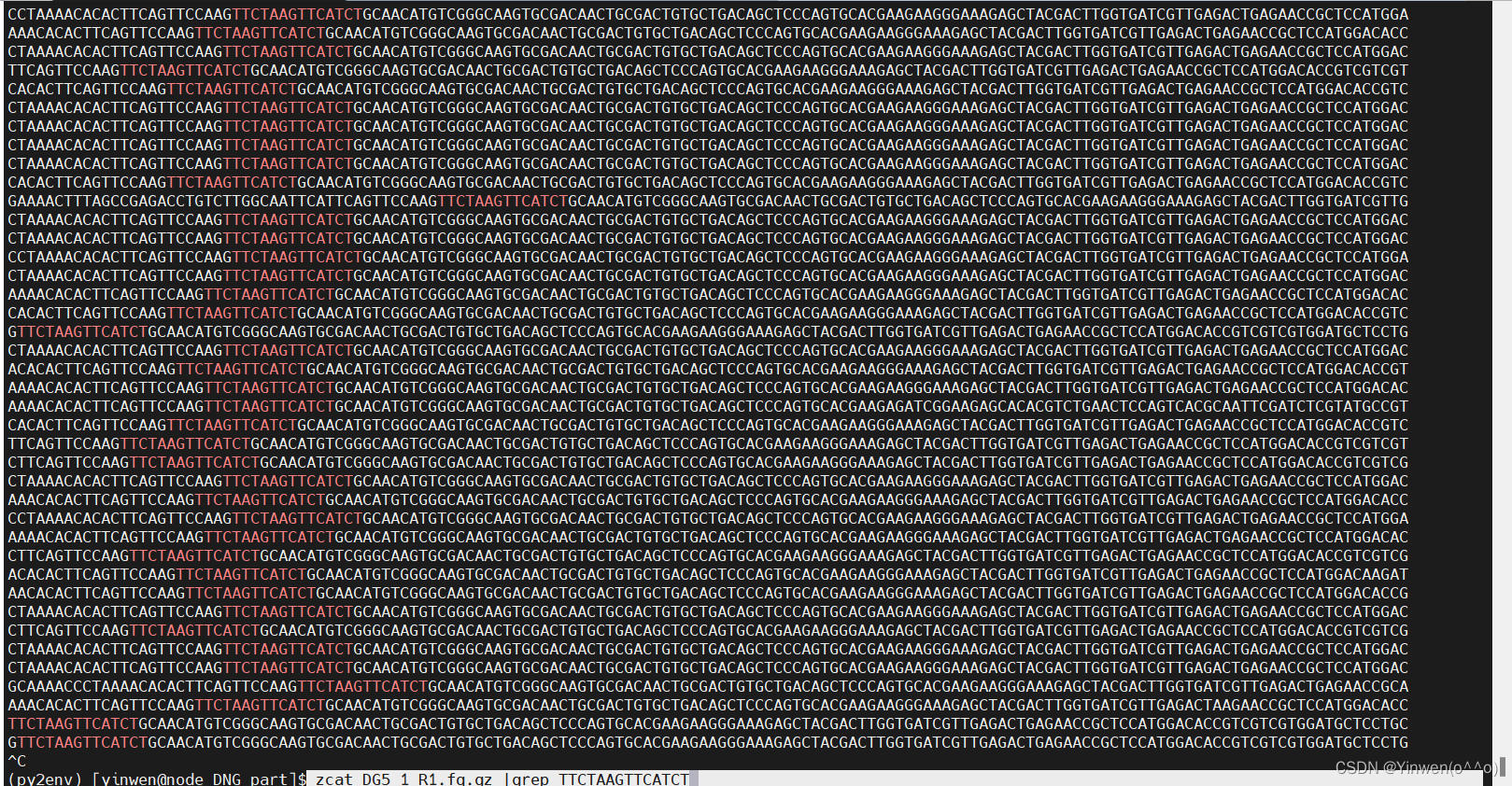
如上图,说明里面含有接头,这些序列是没有意义的,需要过滤
#先用视频里的trim_galore处理单个样本,跑完就是过滤好的数据了
fq1=/home/yinwen/biosoft/DNG_part/DG5_1_R1.fq.gz
fq2=/home/yinwen/biosoft/DNG_part/DG5_2_R1.fq.gz
trim_galore -q 25 --phred33 --length 75 -e 0.1 --stringence 3 --paired --fastqc -o /home/yinwen/clean/ $fq1 $fq2

这里发现跑出来的报告不对,接头还在,序列重复水平依很高,过滤失败。
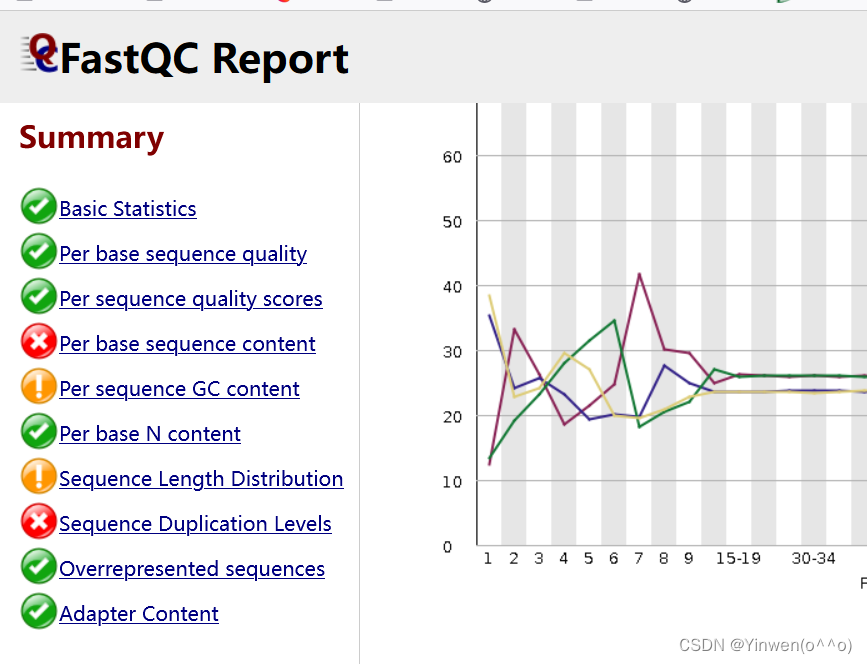
观察模块的结果,为后续过滤,做好准备
观察后发现:
①5’端的碱基,剪掉18bp。
②3’端的碱基不需要修剪,质量很高。
③Adapt接头序列进行修剪。
④重复序列偏多,需要去除。
#修饰参数,5’端的碱基,剪掉18bp,接头内容正常了。
trim_galore -q 20 --phred33 --fastqc --stringency 3 --length 75 -e 0.1 --paired --dont_gzip --clip_R1 18 --clip_R2 18 --illumina -o ./ /home/yinwen/biosoft/DNG_part/DG5_1_R1.fq.gz /home/yinwen/biosoft/DNG_part/DG5_1_R2.fq.gz


#看一下重复序列
zcat DG5_1_R1.fq.gz |grep CCTCGTTCTCGGCGGCAGGAGCATCCACGACGACGGTGTCCATGGAGCGG

#还要继续用其他软件去除那个重复序列??
#决定下载?trimmomatic了
conda install -c bioconda trimmomatic
trimmomatic --help



#部分参数介绍
ILLUMINACLIP: Cut adapter and other illumina-specific sequences from the read.
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 # 切除TruSeq3-PE中提供的Illumina适配器,去接头
SLIDINGWINDOW: Perform a sliding window trimming, cutting once the average quality within the window falls below a threshold.
SLIDINGWINDOW:4:15 #扫描4个碱基宽滑动窗口,当每个碱基的平均质量下降到15以下时进行剪切
LEADING: Cut bases off the start of a read, if below a threshold quality
LEADING:3 # 删除前低质量碱基(低于质量3)
TRAILING: Cut bases off the end of a read, if below a threshold quality
TRAILING:3 # 删除后低质量碱基(低于质量3)
CROP: Cut the read to a specified length
CROP:length #从reads开始开始所要保留的碱基数为length
HEADCROP: Cut the specified number of bases from the start of the read
HEADCROP:12 #删除前12个碱基
MINLEN: Drop the read if it is below a specified length
MINLEN:36 # 上述步骤完成后,删除小于36个碱基的reads (放最后)
TOPHRED33: Convert quality scores to Phred-33
-phred33 #表示将质量分数转换为 Phred-33
TOPHRED64: Convert quality scores to Phred-64
-phred64 #表示将质量分数转换为 Phred-64trimmomatic PE -phred33 DG5_1_R1.fq.gz DG5_1_R2.fq.gz output_forward1__paired.fq.gz output_forward1_unpaired.fq.gz output_reverse2_paired.fq.gz output_reverse2_unpaired.fq.gz ILLUMINACLIP:/home/yinwen/miniconda3/pkgs/trimmomatic-0.39-hdfd78af_2/share/trimmomatic-0.39-2/adapters/TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:20 SLIDINGWINDOW:4:15 HEADCROP:18 MINLEN:75跑完后之前的又又又失败了,显路径不对,我也是用connda安装的,改路径,不报错,再跑。

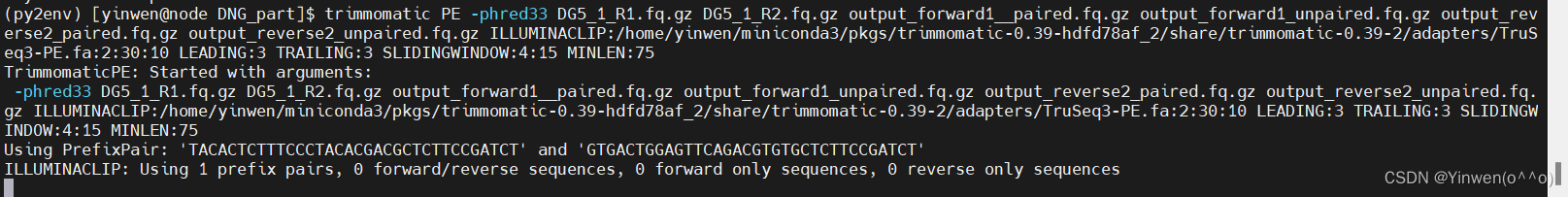

会用trimmomatic了,但是序列重复还是没解决。
对于“Sequence Duplication Levels"中的大量重复我查阅资料发现在单细胞数据中是正常的。
也或许要的就是这个过表达的效果?

我跑出来获得的过表达序列的图:



经过过滤,其reads长度集中分布在120-130nt的位置几平与测序读长一致,且未见明显的小片段reads,表明测序数据质量良好。此处报“WARN”是由于reads长度不一致 (clean reads长度不一完全属正常现象)
?
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!