Nat Cancer | CRISPR-Cas9筛选确定KAT8为PD-L1调控因子
今天给同学们分享一篇实验文章“Disrupting the phase separation of KAT8-IRF1 diminishes PD-L1 expression and promotes antitumor immunity”,这篇文章发表在Nat Cancer期刊上,影响因子为22.7。

结果解读:
CRISPR-Cas9筛选确定KAT8为PD-L1调控因子
考虑到T细胞分泌的IFNγ已被证明是肿瘤微环境的重要调节因子之一,并且是PD-L1的关键和最强诱导因子之一,作者使用全基因组CRISPR-Cas9基因敲除筛选方法,以无偏见的方式鉴定了IFNγ暴露后肿瘤细胞中PD-L1表达的调控因子。已经被确认为PD-L1关键调控因子的一些成熟基因,如JAK1、IFNGR2、STAT1、IRF2以及最近报道的CMTM6和STUB1,在排名靠前的基因中富集。有趣的是,编码组蛋白乙酰转移酶KAT8的基因是作者筛选中富集程度最高的基因之一(图1a、b,扩展数据图图1a)。作为在哺乳动物细胞中催化组蛋白H4赖氨酸16乙酰化(H4K16ac)的主要赖氨酸乙酰转移酶,KAT8还可以乙酰化非组蛋白蛋白质,并在各种细胞过程中发挥重要作用,包括自噬、应激反应和细胞核-线粒体通讯。然而,KAT8在肿瘤进展中的作用以及KAT8如何调节肿瘤免疫微环境仍然定义不清。
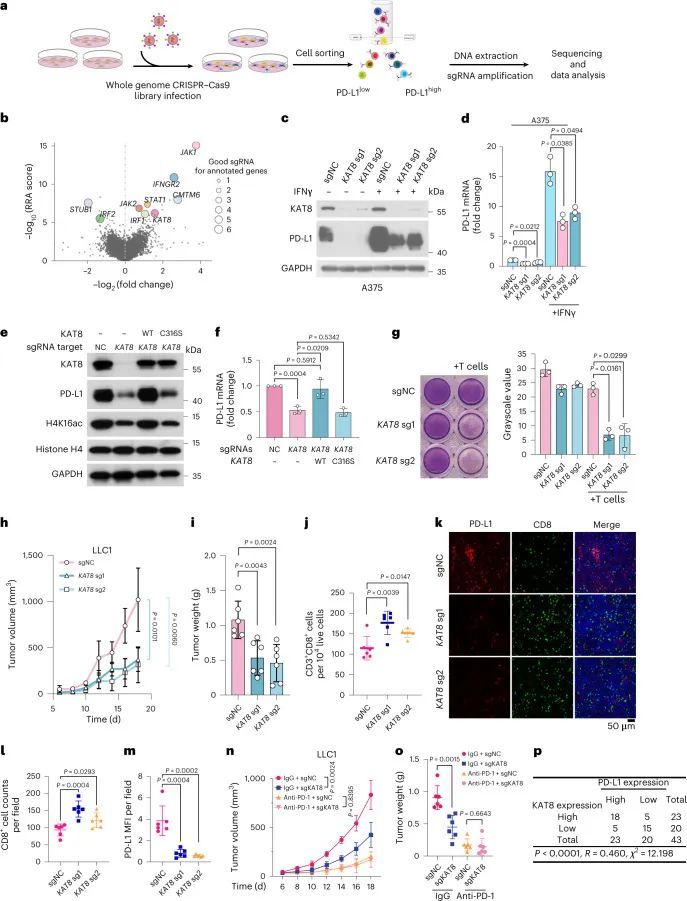
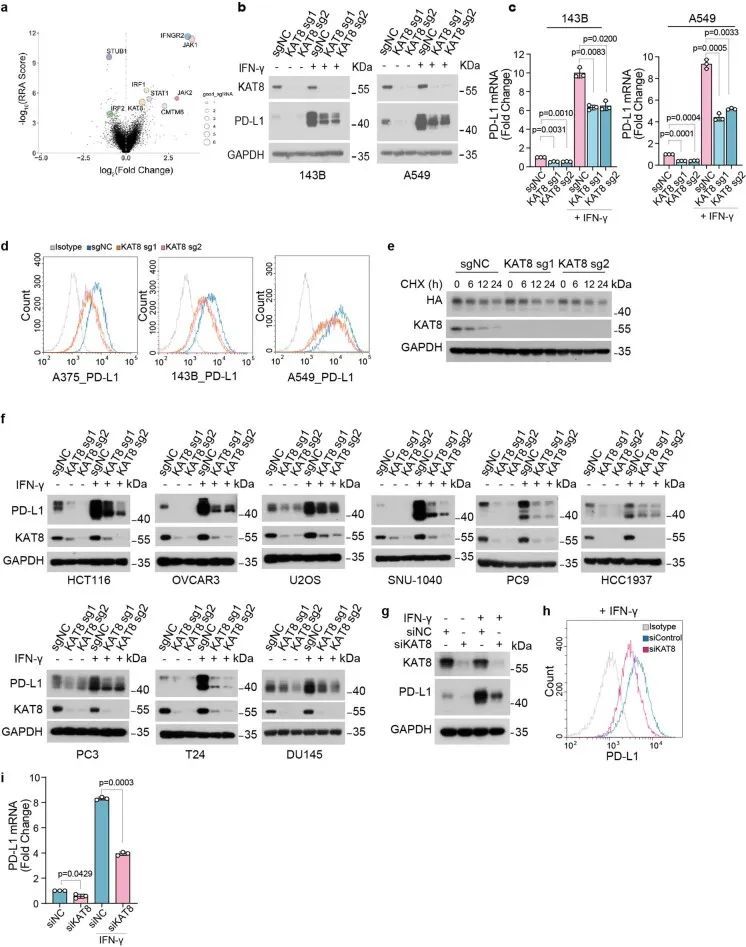
Cas9和单导RNA(sgRNA)在多个细胞系(骨肉瘤细胞系143B、恶性黑色素瘤细胞系A375和肺癌细胞系A549)中异位表达,显著降低了PD-L1的总蛋白质和mRNA水平,无论是否暴露于IFNγ(图1c、d和扩展数据图1b、c)。细胞表面的PD-L1水平也降低了(扩展数据图1d)。此外,KAT8的耗竭并不影响PD-L1蛋白质的半衰期(扩展数据图1e)。这些结果表明,KAT8在各种癌细胞系中调控PD-L1 mRNA的转录。此外,KAT8耗竭引起的PD-L1表达降低在大量细胞系中观察到(扩展数据图1f)。重要的是,野生型(WT)KAT8,而不是其C316S催化缺陷突变体,可以恢复KAT8耗竭细胞中PD-L1和H4K16ac的下调,表明KAT8的乙酰转移酶活性对于调控PD-L1表达至关重要(图1e、f)。同样,使用靶向KAT8的小干扰RNA处理细胞导致PD-L1表达降低(扩展数据图1g-i)。
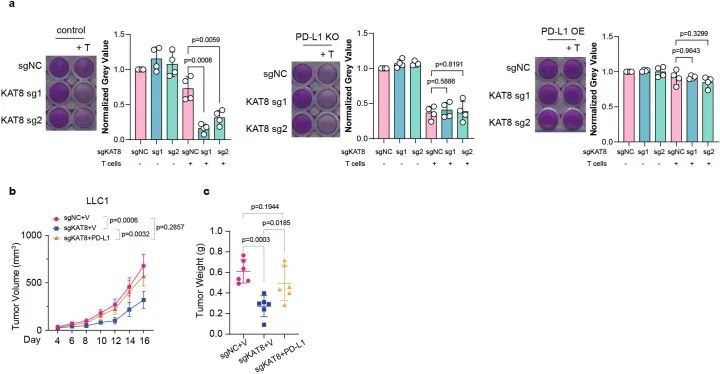
接下来,作者检查了KAT8耗竭对PD-L1表达的下调是否影响抗肿瘤反应。体外细胞毒性实验显示,KAT8耗竭显著增强了T细胞的杀伤能力(图1g),而PD-L1的过表达则逆转了这种增强效应(扩展数据图2a)。在CD247基因敲除细胞中,KAT8耗竭后未观察到进一步的T细胞杀伤增强效应(扩展数据图2a)。在体内,KAT8耗竭抑制了Lewis肺癌细胞系LLC1的肿瘤生长并减轻了肿瘤重量(图1h,i),同时增加了小鼠中CD3 + CD8 + T细胞的肿瘤浸润(图1j-m)。此外,过表达PD-L1可以逆转KAT8耗竭的抗肿瘤效应(扩展数据图2b,c)。在接受抗PD-1治疗的小鼠中,KAT8耗竭无法进一步延缓肿瘤生长(图1n,o)。这些数据表明,KAT8的耗竭通过PD-L1/PD-1轴增强了体外和体内的抗肿瘤免疫力。此外,43个样本中有33个(7674%)人类多器官癌组织阵列样本显示通过免疫组织化学(IHC)检测,KAT8和PD-L1的表达水平呈现高度同步的高或低表达水平(图1p),表明KAT8和PD-L1在人类癌症中存在正相关关系。
KAT8与IRF1相互作用并形成动态凝聚体
为了研究KAT8如何调控PD-L1 mRNA的转录,作者采用了近距离标记方法,将TurboID标记的KAT8引入A375细胞中,标记潜在的相互作用蛋白(图2a)。通过质谱分析,作者确定IRF1是其中一个被标记的蛋白(扩展数据图3a),IRF1已被报道为IFNγ通路中诱导PD-L1 mRNA转录的主要转录因子。事实上,表达靶向IRF1的sgRNA的细胞在IFNγ诱导的PD-L1表达上显示出明显的抑制作用(扩展数据图3b)。通过BirA*近距离标记方法进一步确认了KAT8和IRF1的空间共定位(扩展数据图3c,d)。通过内源性和外源性免疫沉淀进一步确认了KAT8和IRF1之间的相互作用(图2b和扩展数据图3e)。
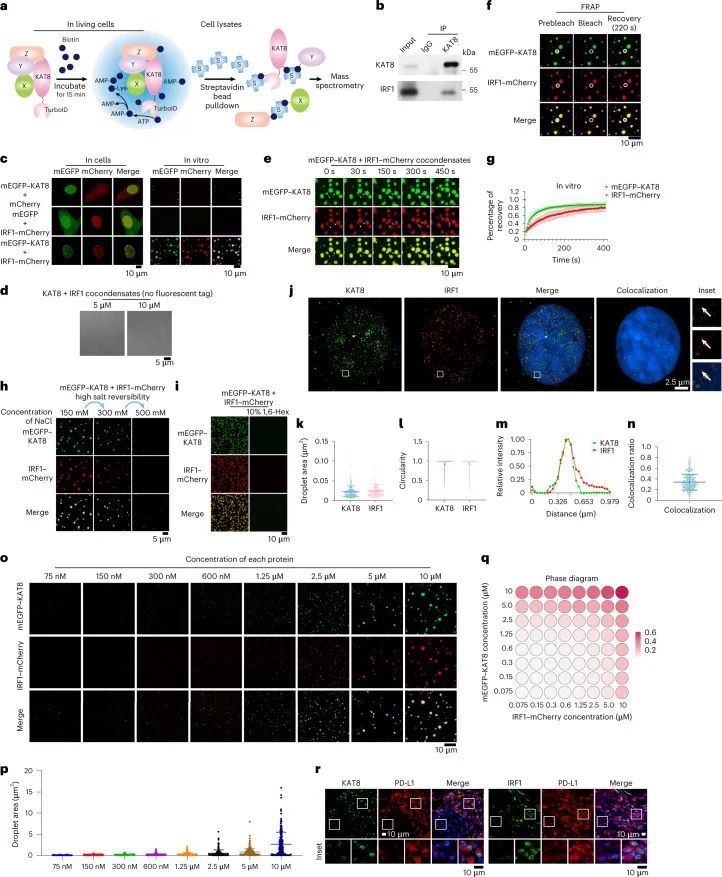
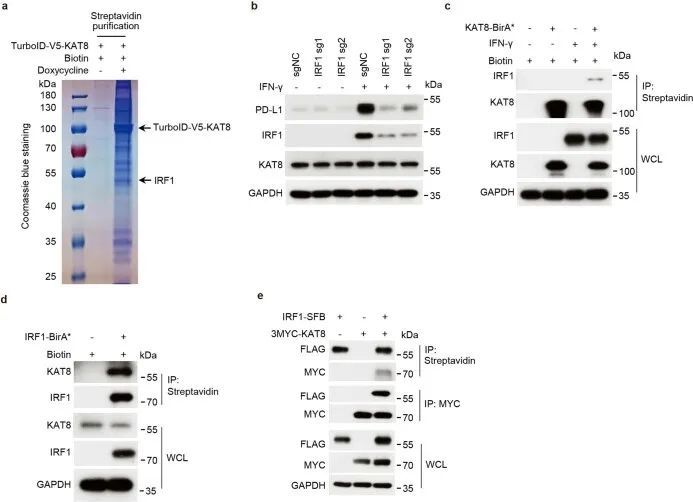
细胞转染了单体增强型绿色荧光蛋白-KAT8(mEGFP-KAT8)和IRF1-mCherry后,在细胞核内形成了类似液滴的凝聚体(图2c,左)。三维结构光学显微镜(3D-SIM)重建也显示了类似液滴的结构(扩展数据图4a)。这些凝聚体对脂质标记染料DiD呈阴性反应(扩展数据图4b),与核仁不共定位,部分共定位于Cajal小体和PML小体(扩展数据图4c),表明这些凝聚体是无膜结构。然后作者研究了KAT8-IRF1凝聚体的动态特性。纯化的mEGFP-KAT8或IRF1-mCherry单独表达时,形成液滴的能力较弱,而两者混合表达则显著增强了液滴的形成(图2c,右,以及扩展数据图4d)。液滴的形成不依赖于荧光蛋白标签,也不依赖于KAT8乙酰转移酶活性(图2d和扩展数据图4e)。此外,滴状物质能够随着时间的推移融合,并在体外和细胞中部分恢复(图2e-g和扩展数据图5a,b);这些滴状物质可以被1,6-己二醇和高浓度的氯化钠破坏(图2h,i)。总的来说,这些结果表明了KAT8-IRF1凝聚物的动态和可逆性质。
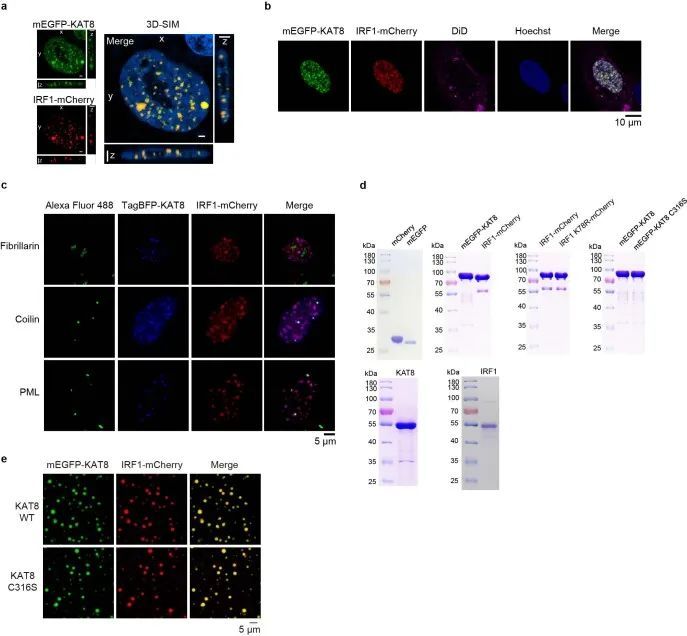
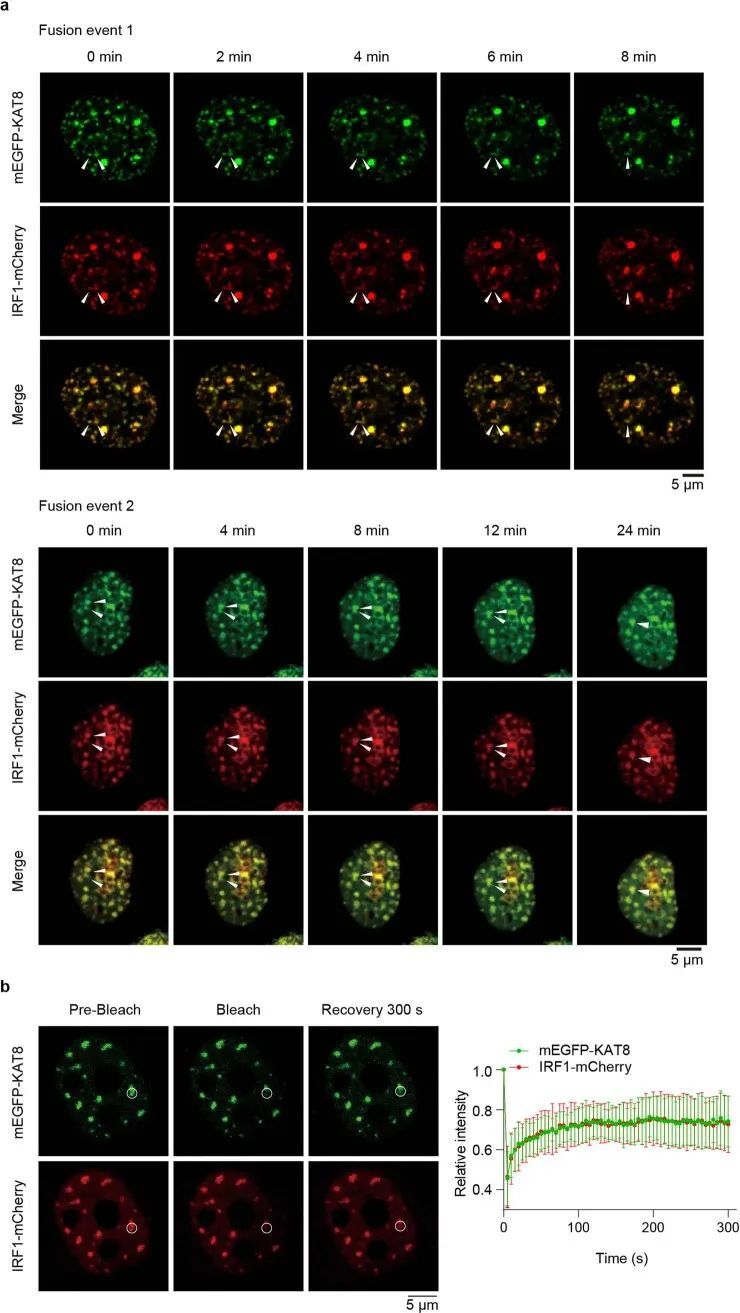
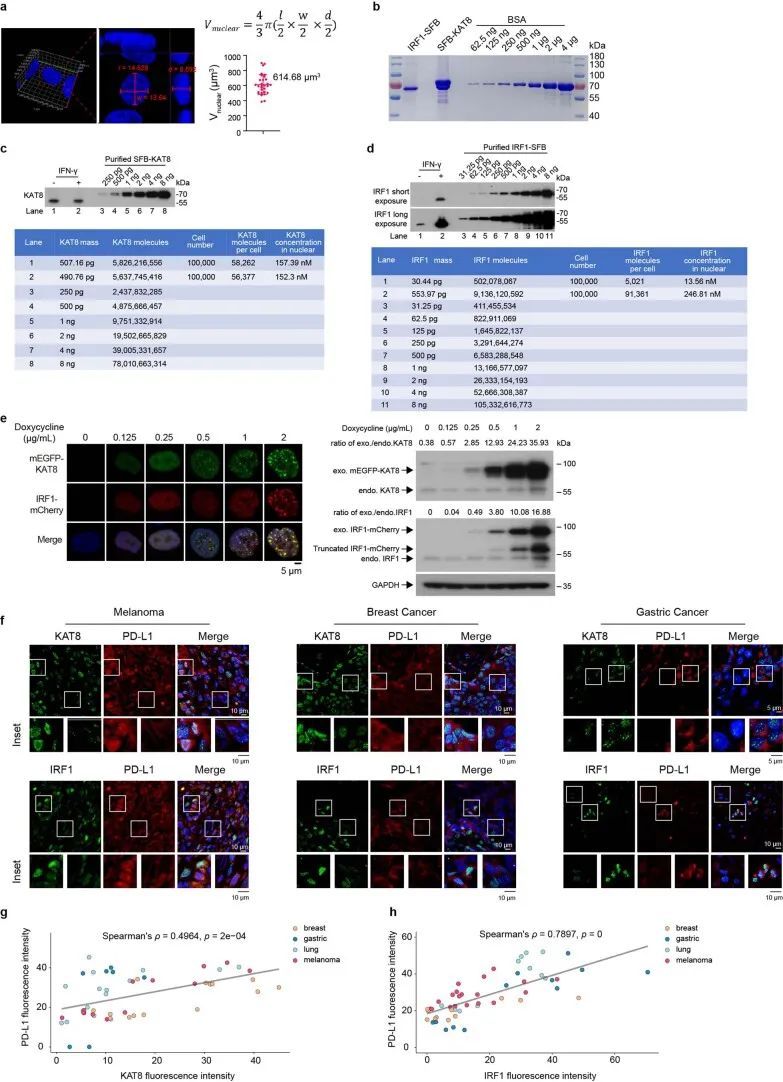
超分辨率成像显示内源性KAT8和IRF1在细胞中形成小的凝聚体,并观察到共定位的凝聚体(图2j-n)。此外,当两种蛋白质浓度高于75 nM时,KAT8-IRF1滴形成于体外(图2o-q);这些浓度与KAT8和IRF1的内源性表达水平相当(经定量的西方印迹和细胞核的三维重建确定,KAT8为152.3 nM,IRF1为246.81 nM,经IFNγ刺激) 33 (扩展数据图6a-d)。此外,作者通过诱导表达系统观察到细胞中外源性mEGFP-KAT8和IRF1-mCherry的浓度依赖性凝聚体形成(扩展数据图6e)。更重要的是,在癌症患者(肺癌、黑色素瘤、乳腺癌和胃癌)的样本中,也观察到KAT8和IRF1的凝聚体,并且两种蛋白质的荧光强度与PD-L1的表达呈正相关(图2r和扩展数据图6f-h)。综上所述,这些结果表明KAT8和IRF1可以在体内和体外形成凝聚体。
KAT8-IRF1的凝聚取决于多价相互作用
为了探索KAT8-IRF1凝聚体的结构基础,作者使用IUPred2分析了这两种蛋白质的氨基酸序列。KAT8的氨基酸1-68(KAT8 1–68 )和包括转录激活结构域的IRF1 140–325 被评分为无序区(IDRs)(图3a,b)。KAT8 1–68 或IRF1 140–325 的消耗破坏了凝聚体的形成(图3c,d),表明预测的IDRs对此过程起到了贡献。然而,共免疫沉淀实验显示,包含N端DNA结合结构域(氨基酸1-115)的IRF1 1–140 ,而不是预测的IRF1 IDR,负责与KAT8的相互作用(图3e),并且IRF1 DBD的缺失(即IRF1 115–325 片段)也影响了与KAT8的凝聚体形成(图3c,d),表明IRF1的DBD介导了与KAT8的相互作用,并且对于凝聚体的形成也是必需的。此外,将IRF1 1–140 与来自不相关转录因子MYC或OCT4的IDRs融合,可以恢复与KAT8的凝聚体形成,而仅使用MYC或OCT4的IDR构建无法实现(图3f)。一个光滴测定法还显示,当KAT8和IRF1的IDR与Cry2融合时,蓝光诱导增强了两种蛋白质的IDR之间的凝聚体形成(图3g,h)。总的来说,这些观察结果表明了一种多价相互作用模型,其中IRF1的DBD和KAT8之间的相互作用可能是凝聚体起始的先决条件,而IRF1和KAT8的IDR之间的弱杂交相互作用促进了凝聚体的形成。
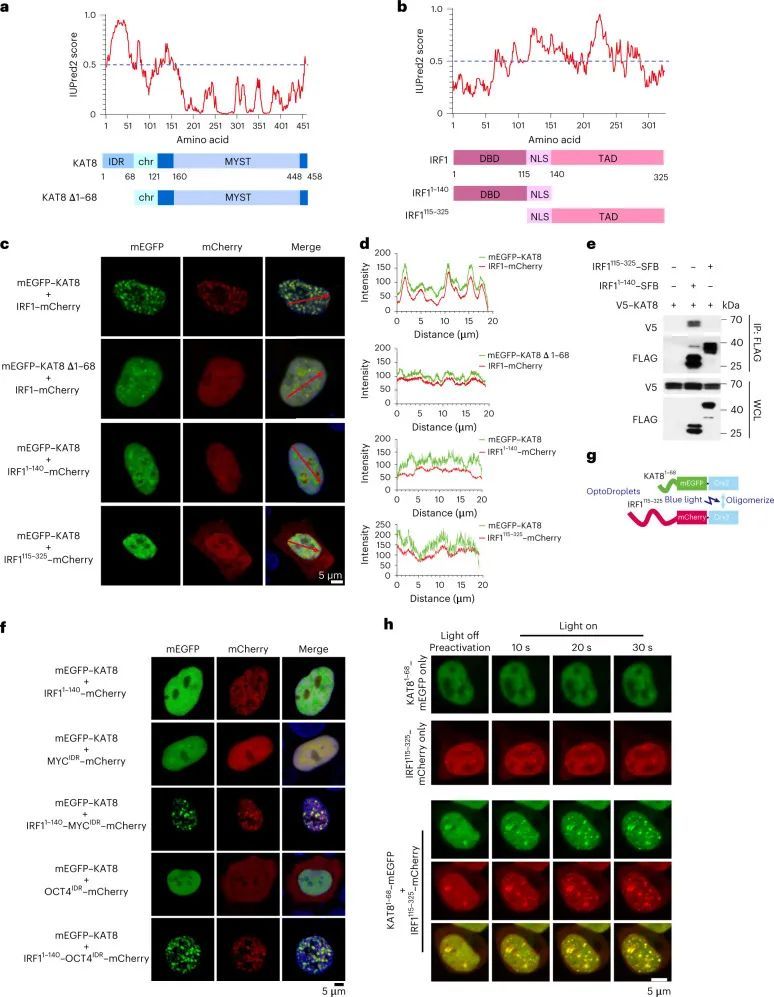
KAT8–IRF1缩合物促进PD-L1 mRNA反式激活
接下来,作者调查了KAT8-IRF1凝聚体是否能增强转录。转录机制的组成部分,包括MED1 IDR、CDK7、CDK9、BRD4和在丝氨酸5位点磷酸化的活性RNA聚合酶II(RNA Pol II-S5P),在KAT8-IRF1凝聚体中富集(图4a,b)。为了测试IDR介导的KAT8-IRF1凝聚在转录增强中的贡献,作者设计了一个拉帕霉素诱导的相互作用系统,通过将KAT8 IDR或IRF1 IDR与FKBP12或FRB-Gal4 DBD融合来解耦结构化的IRF1 DBD-KAT8相互作用和杂交的IDR相互作用。拉帕霉素处理后,共转染KAT8 IDR-FKBP12和IRF1 IDR-FRB-Gal4 DBD的细胞在细胞核中显示出小的凝聚体(图4c),表明该系统可以模拟由KAT8和IRF1 IDR相互作用引起的凝聚。然后,作者使用Gal4上游激活位点(UAS)报告基因分析评估了IDR凝聚的转录激活效应(图4d)。转染IRF1 IDR-FRB-Gal4 DBD增加了报告基因的表达,而无IDR对照和KAT8 IDR-FKBP12没有活性,并且拉帕霉素诱导不能进一步增强报告基因的表达。细胞共转染了KAT8 IDR-FKBP12和IRF1 IDR-FRB-Gal4 DBD后,在没有雷帕霉素的情况下,显示出与仅转染IRF1 IDR-FRB-Gal4 DBD的细胞相似的报告基因表达水平,而在雷帕霉素处理后,报告基因表达显著增强(图4e)。此外,KAT8 IDR-FKBP12和IRF1 IDR-FRB-Gal4 DBD稳定整合的UAS报告基因细胞中,雷帕霉素剂量-反应曲线显示出非线性激活模式(图4f)。这些结果表明,IDR介导的KAT8-IRF1凝聚促进了转录激活。此外,在进行dCas9-SunTag-sgARRAY介导的原位标记后,作者观察到内源性KAT8和IRF1在细胞中在PD-L1启动子处形成凝聚体(图4g-k)。
已经证明KAT8在男性特异性致死(MSL)和非特异性致死(NSL)复合物中充当催化核心亚单位,这些复合物对组蛋白和非组蛋白底物具有不同的催化活性。作者的数据显示,MSL和NSL复合物亚单位也参与了KAT8-IRF1凝聚体的形成(扩展数据图7a,b)。
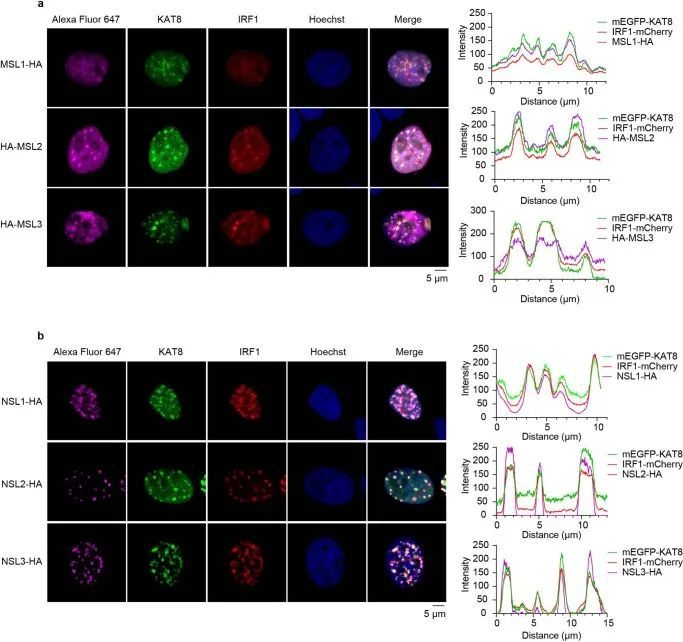
KAT8乙酰化并促进IRF1活性
接下来,作者测试了KAT8是否能够乙酰化IRF1。在使用组蛋白去乙酰转移酶抑制剂三氢史坦A(TSA)和烟酰胺(扩展数据图8a)处理后,检测到IRF1的乙酰化并得到增强。与IRF1共转染野生型KAT8(WT KAT8)相比,其催化活性缺陷的C316S突变体并不能显著增强IRF1的乙酰化(图5a)。通过质谱分析,作者确定了IRF1的七个乙酰化赖氨酸残基(K43、K66、K70、K78、K117、K275和K299;扩展数据图8b)。然后,作者通过将每个确定的乙酰化赖氨酸替换为精氨酸构建了IRF1的单点突变体。当与WT KAT8共转染时,只有IRF1 K78R突变体没有显示出增强的乙酰化(扩展数据图8c)。重要的是,IRF1 K78乙酰化(IRF1 K78ac)是由KAT8特异性催化的,而不是其他组蛋白乙酰转移酶(扩展数据图8d)。在应用特异性抗体对乙酰化的IRF1 K78(K78ac)后,通过体内和体外的乙酰化实验进一步确认了WT KAT8的直接催化活性,而C316S突变体则没有(图 5b、c和扩展数据图8e)。更重要的是,耗竭KAT8导致内源性IRF1在K78位点的乙酰化减少(图5d)。综上所述,这些数据表明KAT8特异性乙酰化IRF1的K78位点。
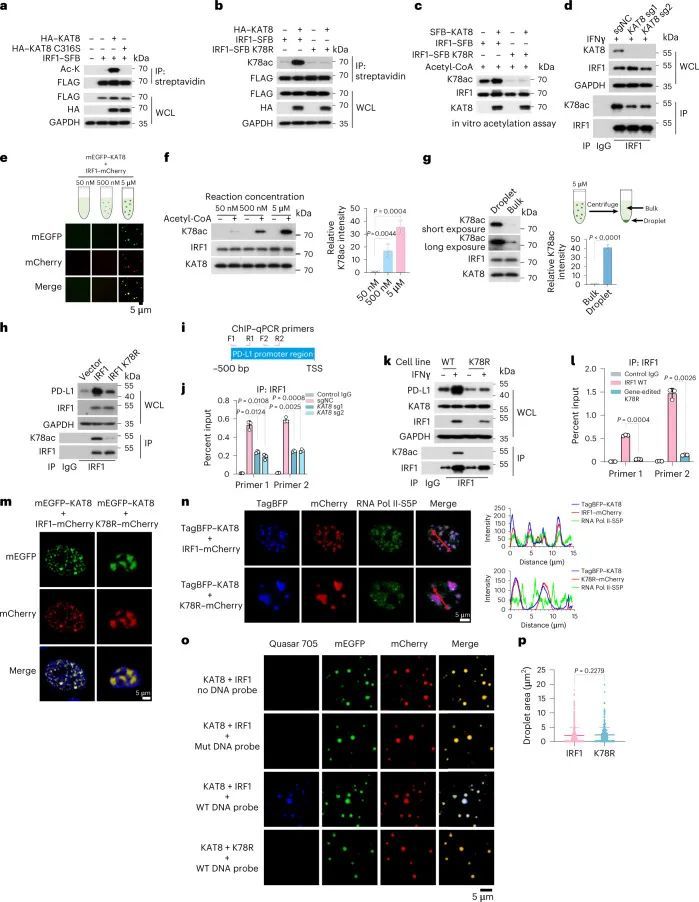
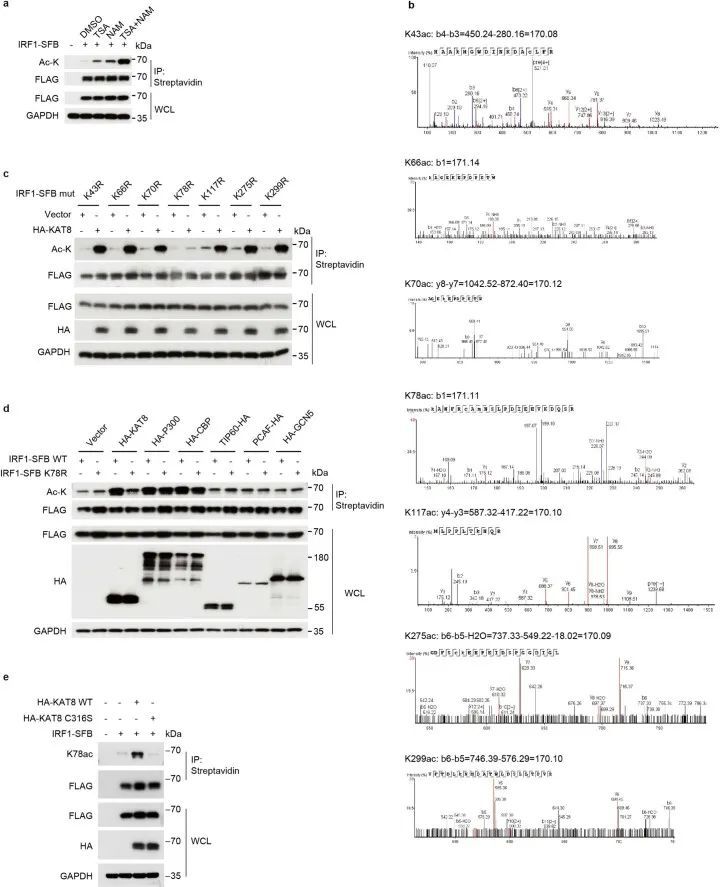
生物分子凝聚物的形成被用作增加酶催化反应速率的机制,因为酶和底物被浓缩在液滴中。为了探索KAT8-IRF1凝聚物是否促进IRF1 K78ac,进行了带有或不带有液滴形成的体外组蛋白乙酰转移酶活性测定。在液滴中,IRF1 K78ac显著增加(图5e,f),而液滴中KAT8的乙酰化能力约为批量状态下的39.67±1.997倍(图5g)。这些结果提供了KAT8通过共凝聚促进IRF1乙酰化的证据。
接下来,作者评估了IRF1 K78ac对PD-L1表达的影响。IRF1 K78R突变体未能上调PD-L1表达,并且在PD-L1启动子上显示出减少的丰度(图5h和扩展数据图9a)。此外,IRF1同源二聚体化对IRF1 K78的乙酰化没有影响(扩展数据图9b)。这些结果表明,K78的乙酰化对IRF1的DNA结合很重要。与这些数据一致,通过sgRNA处理使KAT8耗尽显著减少了PD-L1启动子上IRF1的丰度(图5i,j)。为了进一步支持这个结论,作者使用CRISPR-Cas9介导的同源定向修复技术在A375细胞系中生成了一个特定位点的K78R敲入细胞系(以下简称K78R细胞)(扩展数据图9c-e)。K78R细胞的PD-L1 mRNA和蛋白表达显著降低(图5k和扩展数据图9f)。在经过IFNγ处理的K78R细胞中,PD-L1启动子上IRF1的丰度也显著降低(图5l)。KAT8-IRF1 K78R形成的细胞内凝聚体一贯地显示出大型不规则和较少动态的聚集,与RNA Pol II-S5P不共定位(图5m,n和扩展数据图9g,h)。虽然K78R突变体在体外与KAT8相互作用和共凝聚的能力类似(图5o,p和扩展数据图9i),但KAT8-IRF1 K78R形成的液滴与来自PD-L1启动子的DNA探针的招募显著减少,与WT液滴相比(图5o)。综上所述,这些结果表明,KAT8在K78位点乙酰化IRF1,增强了IRF1的DNA结合活性以及其定位到PD-L1启动子和随后的PD-L1 mRNA转录的激活。
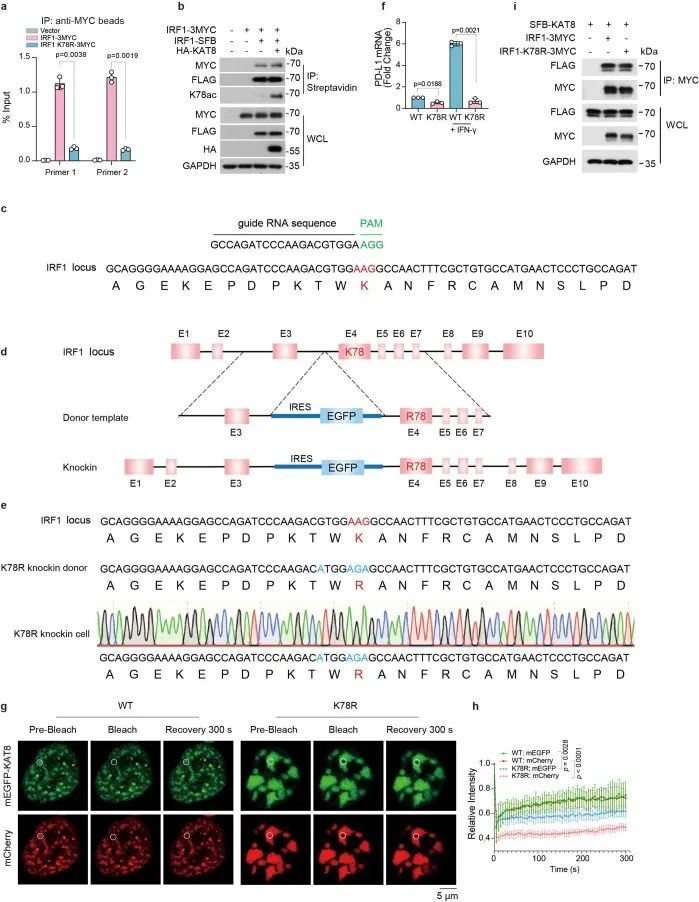
IRF1乙酰化将KAT8招募到PD-L1启动子
鉴于KAT8是哺乳动物细胞中H4K16ac的主要乙酰转移酶,作者调查了KAT8是否调控PD-L1启动子中的H4K16ac。表达针对KAT8的sgRNA的细胞显示H4K16ac总体减少,而其他H4乙酰化位点(H4K5ac、H4K8ac和H4K12ac)保持不变(图6a)。来自ENCODE数据库的染色质免疫沉淀测序(ChIP-seq)数据显示KAT8和IRF1富集在PD-L1的启动子区域(参考文献,图6b)。ChIP-定量PCR(ChIP-qPCR)显示当KAT8表达受sgRNA抑制时,PD-L1启动子中的H4K16ac也显著减少(图6c)。
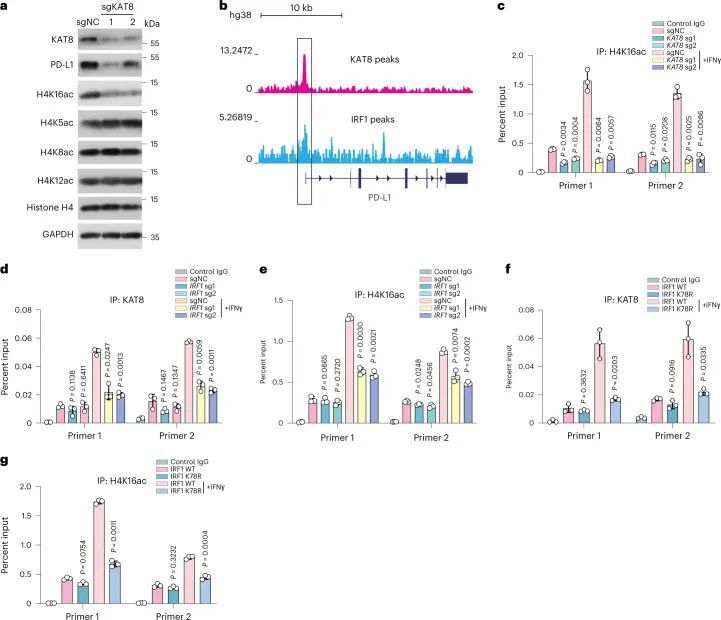
破坏KAT8-IRF1凝聚体抑制PD-L1表达
考虑到KAT8-IRF1凝聚体在调控PD-L1转录中的作用,作者推测破坏KAT8-IRF1的相分离可能会抑制PD-L1 mRNA的转录。删除IRF1 DBD导致凝聚体形成受损(图3c,d),而将两种蛋白质的IDR与Cry2(蓝光诱导的相互作用;图3g,h)或FKBP12-FRB(雷帕霉素诱导的相互作用;图4c,d)融合增强了IDR的凝聚体形成。这些结果突出了IRF1 DBD和KAT8之间结构域相互作用在凝聚体形成中的重要作用,表明与目标为非结构化IDR相比,目标为结构域相互作用也可能破坏KAT8-IRF1凝聚体。为了验证这个假设,作者构建了一系列IRF1 DBD截断(图7a),并将每个截断与KAT8共转染到细胞中,以确定IRF1与KAT8相互作用的最小区域。完全保守的N末端区域(氨基酸21-42,以下简称2142)在人类和小鼠中被确定为IRF1-KAT8相互作用的主要区域,其中包含一个双链β-折叠结构 36 ,并且预测的β-折叠关键残基的突变(Mut)会消除这种相互作用(图7c-f)。
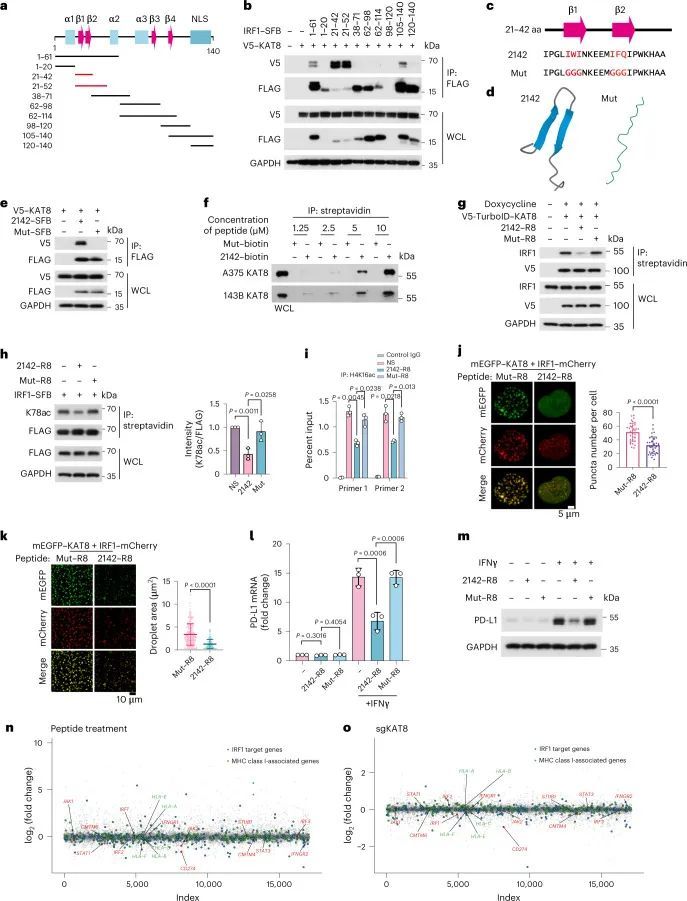
接下来,作者合成了两种肽,2142–R8和Mut–R8,来源于2142和Mut,其C末端(R8)融合有八个精氨酸残基,增强了细胞膜渗透和核定位(扩展数据图10a)。肽可以进入细胞核并具有低细胞毒性(扩展数据图10b,c)。为了评估2142–R8在细胞生理条件下对KAT8–IRF1缩合物的破坏能力,使用了带有V5 TurboID标记的KAT8的邻近标记系统(图2a)。用2142–R8处理细胞后,生物素标记的IRF1水平显著降低(图7g)表明细胞中IRF1和KAT8之间的相互作用被抑制。因此,IRF1 K78的乙酰化被抑制,PD-L1启动子处的H4K16ac被减少(图第7h,i段)。一致地,在细胞和体外2142–R8处理后,KAT8–IRF1缩合物减少(图第7j,k段)。最重要的是,2142–R8在用IFNγ处理的细胞中有效抑制了PD-L1表达(mRNA和蛋白质)的上调(图7l,m),但未能进一步降低表达靶向KAT8的sgRNA的细胞或基因编辑的K78R细胞中的PD-L1 mRNA和蛋白质水平(扩展数据图10d–g),表明2142–R9对PD-L1的抑制取决于KAT8–IRF1的相互作用。此外,RNA测序分析显示,2142–R8处理下调了PD-L1,而主要组织相容性复合体I类(MHC I类)相关基因和大部分IRF1下游的表达保持不变,这与KAT8缺失细胞的数据一致(图7n、o)。
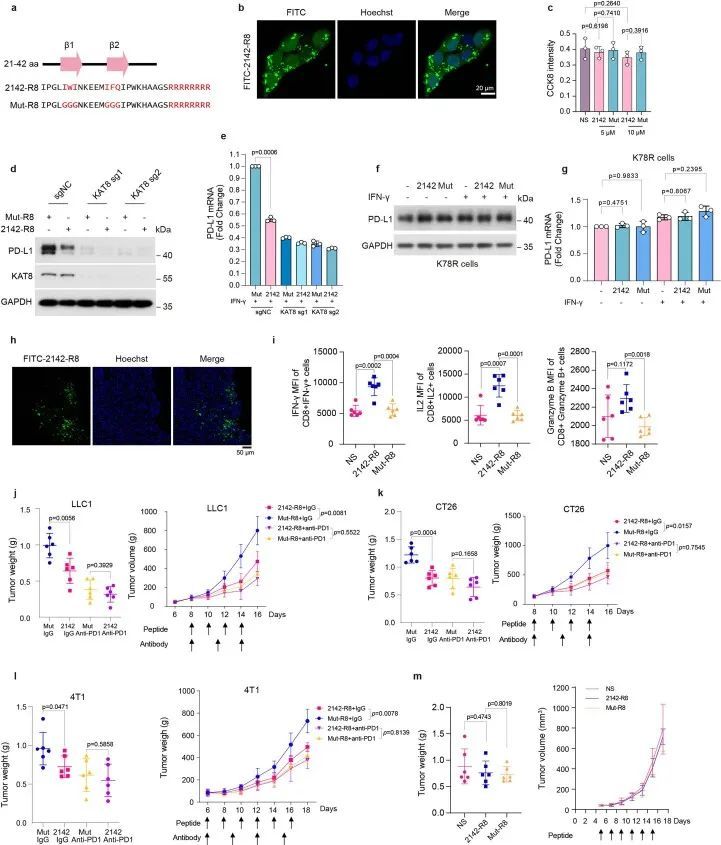
2142–R8肽增强抗肿瘤免疫力
然后,作者评估了2142–R8在增强抗肿瘤免疫方面的功效。体外细胞毒性测定显示,2142–R8(而不是Mut–R8)增强了T细胞杀伤(图8a)。在体内,腹膜内注射肽可以渗透到肿瘤组织中(扩展数据图10h)。用2142–R8而不是Mut–R8治疗,降低了携带LLC1肿瘤的小鼠中KAT8–IRF1点状点的共定位、肿瘤体积和肿瘤重量(图8b–e)。同样,肿瘤PD-L1表达减少,活性CD8+T细胞的浸润增加2142–R8,但不增加Mut–R8(图8f–l)。2142–R8处理还增强了小鼠肿瘤中浸润的CD8+T细胞的活化(扩展数据图10i)。2142–R8的肿瘤抑制作用也在CT26和4T1肿瘤模型中观察到(扩展数据图10j–l)。相反,2142–R8在通过抗PD-1治疗阻断PD-1/PD-L1轴时或在NOG小鼠中未能进一步增强肿瘤减少(扩展数据图10j–m)。总之,这些结果表明2142-R8肽在体内抑制PD-L1的表达并增强抗肿瘤免疫。
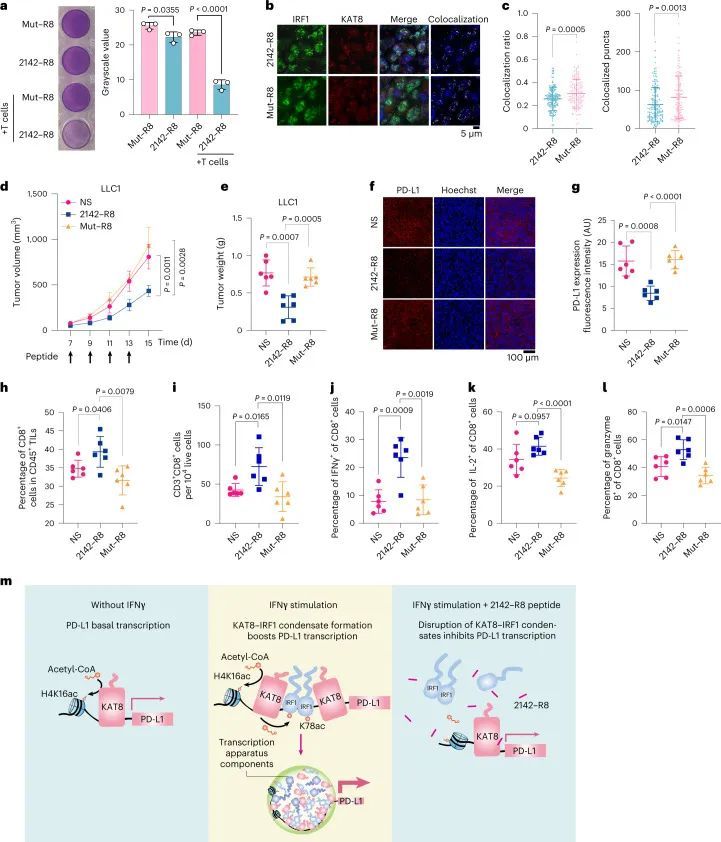
总结
癌症发展的调控中越来越多的证据表明生物分子凝聚体的作用,这表明针对这一过程可能是有希望的。在KAT8-IRF1凝聚体的背景下,作者证明了IRF1 DBD和KAT8之间的特定结构化相互作用是凝聚体形成的先决条件,而IRF1和KAT8 IDRs的弱杂乱相互作用促进了凝聚体的形成。基于这一机制,作者发现了2142-R8肽,它可以阻断IRF1 DBD和KAT8的相互作用,破坏KAT8-IRF1凝聚体的形成,进而抑制PD-L1的表达,增强体外和体内的抗肿瘤免疫。总的来说,作者的数据表明抑制与癌症相关的凝聚体形成可能是癌症治疗的潜在策略。对这篇文章的思路感兴趣的老师,欢迎咨询!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- 细说JavaScript语句详解
- 一篇文章掌握 NestJS 所有的生命周期以及生命周期的执行时机
- 网络安全(黑客)—2024自学
- Python 与 PySpark数据分析实战指南:解锁数据洞见
- Liunx最详细基础命令
- 机器学习之集成学习AdaBoost
- windows系统安装RocketMQ_dashboard
- 用java以数组为底层结构创建循环队列
- Whisper: openAI开源准确率最高的通用语言语音识别
- Unity中URP下实现深度贴花(雾效支持和BRP适配)