噬菌体序列分析工具PhaVa的使用和使用方法
?挺简单的,这里就不翻译了,大家看着直接用吧。
PhaVa
PhaVa is an approach for finding potentially?Phase?Variable invertible regions, also referred to as invertons, in long-read seqeuncing data
Dependencies
Versions listed are the versions PhaVa has been tested on.
PhaVa is developed and tested on Linux operating systems (CentOS Linux 7), however it should compatible with Mac OSX and Windows
Usage
The PhaVa workflow is divided into three steps: locate, create, and ratio.
phava locate -i genome.fasta -d out_dir
phava create -d out_dir
phava ratio -r long_reads.fastq -d out_dir
Alternatively, all three steps can be run in a single command via variation_wf
phava variation_wf -i genome.fasta -r long_reads.fastq -d out_dir
Output from each step is centered around a output directory (-d) and should be the same directory for the entire workflow The locate and create steps only need to be performed once for a given genome or metagenome, and ratio can then be run on long-read samples using the same output directory (-d)
Any invertons with at least 1 read aligning in the reverse orientation will be found in the output. However, it is strongly recommended to fruther filter based on a minimum reverse read count and minimum % reverse of all reads cutoff (3 and 3% are recommended, respectively)
Expected output:?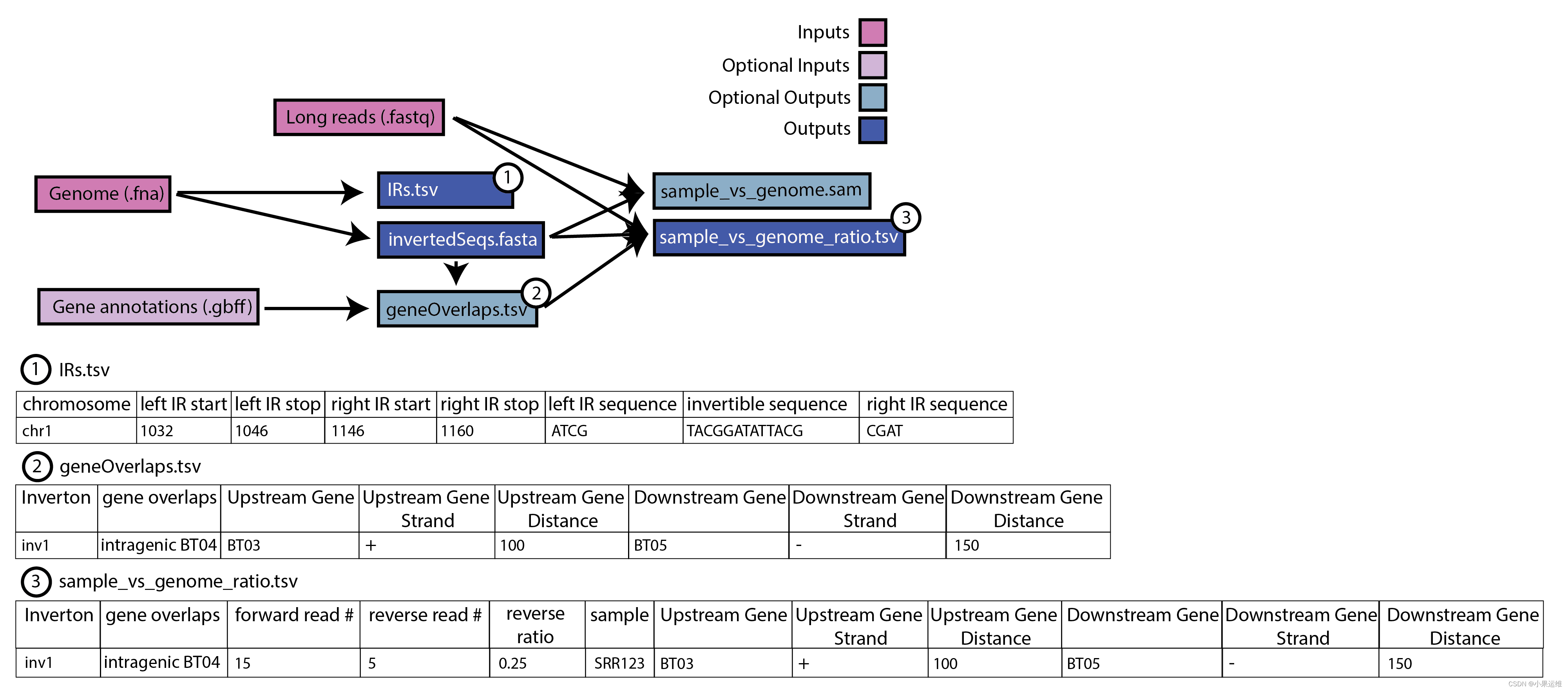
Installation
Beyond installing dependencies, PhaVa install is:
git clone https://github.com/patrickwest/PhaVa
Testing
PhaVa install can be tested on a small simulated dataset, typically in <1 minute, with pytest and a pytest module located in the 'tests' subdirectory:
pytest phava_test.py本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- 介绍 Docker 的基本概念和优势,以及在应用程序开发中的实际应用。
- C++中用于精度控制的在1e-6 的含义
- 华为OD机试真题-中文分词模拟器-2023年OD统一考试(C卷)
- 【Git】Git基本操作
- 【正点原子STM32连载】 第四十四章 外部SRAM实验 摘自【正点原子】APM32E103最小系统板使用指南
- 软件测试/测试开发丨Pytest测试用例生命周期管理-Fixture
- c/c++ 文件操作(2)
- 架构设计到底是什么?
- 《PCI Express体系结构导读》随记 —— 第I篇 第1章 PCI总线的基本知识(6)
- [设计模式Java实现附plantuml源码~创建型] 对象的克隆~原型模式