Trinity软件对转录组进行无参比对教程
写在前面
2023年将结束,小杜的生信笔记分享个人学习笔记也有2年的时间。在这2年的时间中,分享算是成为工作、学习和生活中的一部分。自己为了运行和维护社群也算花费大量的时间和精力,自己认为还算满意吧。对于个人来说,自己一直的目的都是分享自己的学习笔记,以及多多少少可以帮助解决一下需要帮助的同学。我们这里所说的是需要帮助的人,而不是大部分人。自己的能力和精力自己清楚,自己研究的方向也是比较局限,我们并不知专职做这块的博主,等等…。因此,我这边一直在鼓励大家投稿,但事与愿违的事情很多…。
害!最后,还是那句话:一边学习,一边总结,一边分享!
转录组无参比对教程
当作物是没有参考基因组时,需要无参进行比对。Trinity是现在使用最广泛的转录组De novo组装软件。

Trinity 是无参考转录组从头组装转录组的常用软件,且trinity的使用文档非常详细,整合的内容非常完整,包括从组装,比对,定量到差异分析等。因此有大神也推荐Trinity可作为初学者了解熟悉转录组分析流程的入门和进阶学习文档。
–
原文链接:转录组无参比对教程
1.1 软件安装
**官方文档:**https://github.com/trinityrnaseq/trinityrnaseq/wiki
Trinity通过有秩序的对大规模的RNA-seq Reads数据进行读取,高效的完成转录组的组装,包含三个独立的软件模块:
Inchworm
将RNA-seq原始数据组装成unique序列
Chrysalis
将Inchworm 生成contigs聚类,每个类构建Bruijn图
Butterfly
处理Bruijn图,依据图中reads
- conda安装
## 搜索conda的版本
$ conda search trinity
#---
trinity 2.9.1 h8b12597_1 anaconda/cloud/bioconda
trinity 2.11.0 h5ef6573_0 anaconda/cloud/bioconda
trinity 2.11.0 h5ef6573_1 anaconda/cloud/bioconda
trinity 2.12.0 h5ef6573_0 anaconda/cloud/bioconda
trinity 2.12.0 ha140323_1 anaconda/cloud/bioconda
trinity 2.12.0 ha140323_2 anaconda/cloud/bioconda
trinity 2.12.0 ha140323_3 anaconda/cloud/bioconda
trinity 2.13.2 h00214ad_1 anaconda/cloud/bioconda
trinity 2.13.2 h15cb65e_2 anaconda/cloud/bioconda
trinity 2.13.2 ha140323_0 anaconda/cloud/bioconda
trinity 2.13.2 hea94271_3 anaconda/cloud/bioconda
#-----------
conda install -y trinity
- 源码安装
The Trinity software package can be downloaded here on GitHub. Legacy versions (pre-2015) are still available at our Sourceforge Trinity software archive.
Runtime and transcript reconstruction performance stats are available for current and previous releases.
wget https://github.com/trinityrnaseq/trinityrnaseq/archive/refs/tags/Trinity-v2.15.0.zip
unzip Trinity-v2.15.0.zip
##
echo 'PATH=$PATH:~/software/trinityrnaseq-Trinity-v2.15.0'
1.2 Trinity使用
Trinity组装原理
Trinity组装依据的算法是de Bruijn Graph,即从打断的文库中提取一定长度的K-mer,然后根据k-1错位相似的方法拼接组装的可能路径,最终确定完整的参考组装转录组。
Trinity根据该原理,将主要操作步骤分为3个模块,分别形象的命名为虫,蛹,蝶:
- 序列延伸 (inchworm) ——虫
- 将 reads切为 k-mers (k bp长度的短片段)
- 利用Overlap关系对k-mers进行延伸 (贪婪算法)
- 输出所有的序列 (“contigs”)
- 构建 de Bruijn graph (chrysalis)——蛹
- 聚类所有相似区域大于k-1bp的 contigs
- 构图 (区分不同的 “components”)
- 将reads比对回 components,进行验证
- 解图,列举转录本 (butterfly)——蝶
- 拆分graph 为线性序列
- 使用reads以及 pairs关系消除错误序列
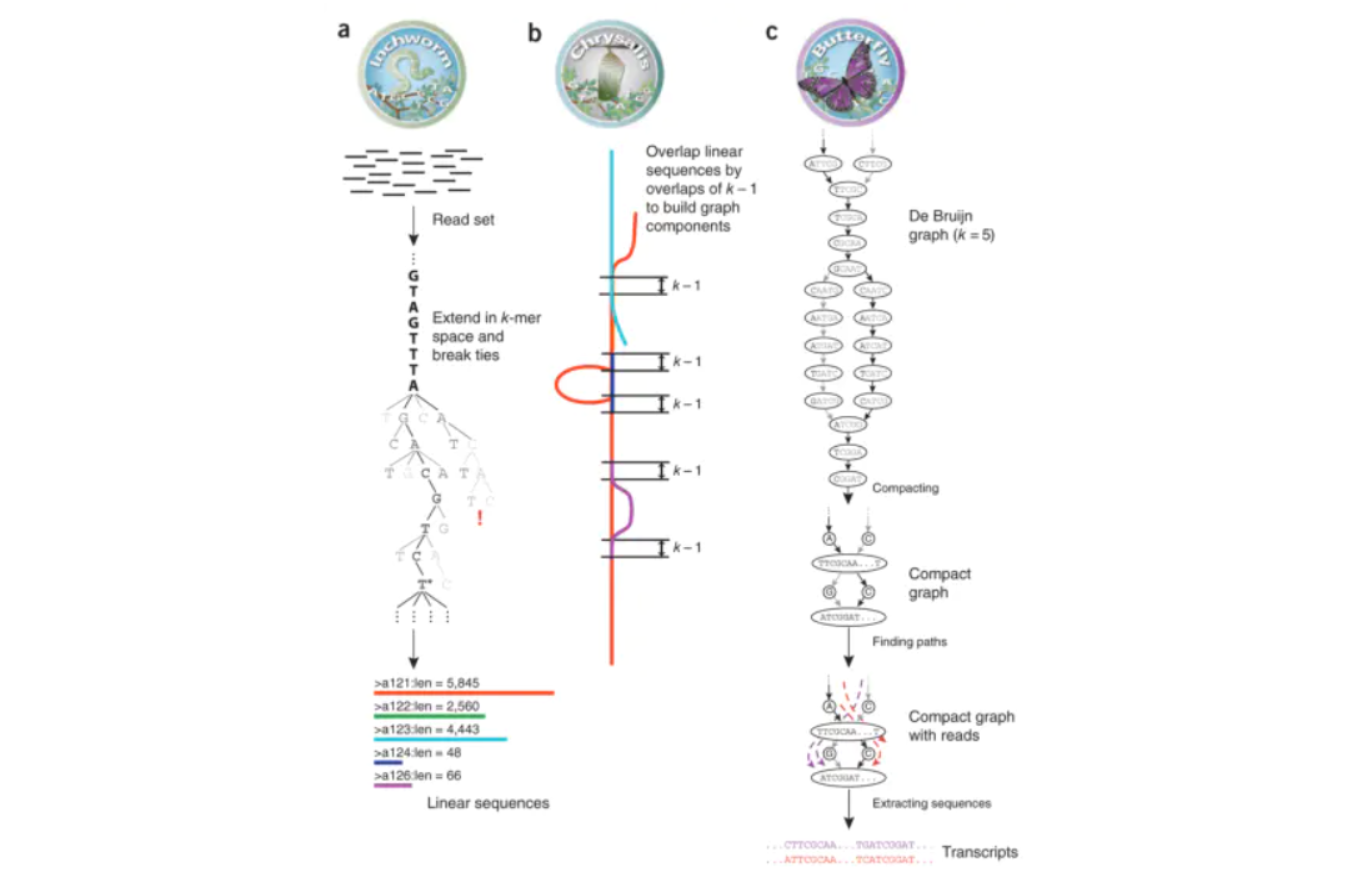
Trinity组装
Trinity --seqType fq --max_memory 100G --left reads_1.fq.gz --right reads_2.fq.gz --SS_lib_type RF --CPU 30 --output ../outputPATH --min_contig_length 200 --jaccard_clip --trimmomatic --normalize_reads --bflyCalculateCPU
必须参数:
--seqType <string> :type of reads: ('fa' or 'fq')
reads的类型
--max_memory <string> :suggested max memory to use by Trinity where limiting can be enabled. (jellyfish, sorting, etc)
provided in Gb of RAM, ie. '--max_memory 10G'
最大内存的大小,GB
--left <string> :left reads, one or more file names (separated by commas, no spaces)
双段转录组数据编号为1的数据,如果对多组数据进行分析,则使用都好`,`将文件进行分开
--right <string> :right reads, one or more file names (separated by commas, no spaces)
双段转录组数据编号为2的数据,如果对多组数据进行分析,则使用都好`,`将文件进行分开
## 或是使用下面的表达方式
or, if unpaired reads:
--single <string> :single reads, one or more file names, comma-delimited (note, if single file contains pairs, can use flag: --run_as_paired )
Or,
--samples_file <string> tab-delimited text file indicating biological replicate relationships.
ex.
cond_A cond_A_rep1 A_rep1_left.fq A_rep1_right.fq
cond_A cond_A_rep2 A_rep2_left.fq A_rep2_right.fq
cond_B cond_B_rep1 B_rep1_left.fq B_rep1_right.fq cond_B cond_B_rep2 B_rep2_left.fq B_rep2_right.fq
# if single-end instead of paired-end, then leave the 4th column above empty.
可选参数:
--SS_lib_type
reads的方向,成对的reads:RF or FR; 不成对的reads:F or R。在数据具有特异性的时候,设置参数,则正义与反义转录子能得到区分。默认情况下,不设置此参数,reads被当做非特异性处理。
RF:reads.1.fq文件的序列和基因序列反向互补,reads.2.fq文件的序列和基因序列一致,次情况下特异性测序的类型。
FR:与RF相反,reads。1.fq文件的序列和基因序列一致,reads。2.fq文件的序列和基因序列互补。
.......
原文链接:转录组无参比对教程
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
4. 精美图形绘制教程
5. 转录组分析教程
一个转录组上游分析流程 | Hisat2-Stringtie
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- JS中map()、forEach()、filter()函数
- 如何传承中国文化
- model.named_children()函数的用法
- uniapp h5 生成 ubuntu桌面程序 并运行方法
- 快递打单软件使用教程
- 智能优化算法应用:基于猎食者算法3D无线传感器网络(WSN)覆盖优化 - 附代码
- 【Redis缓存数据库】Redis的引入及NIO的原理
- 计算机体系结构超标量及分支预测学习记录
- 开启Android学习之旅-6-实战答题App
- 高分辨大尺寸图像的目标检测切图处理