测序名词解释
测序深度(Sequencing Depth)是指:测序得到的碱基总量(bp)与基因组(转录组或测序目标区域大小)的比值,是评价测序量的指标之一。
测序深度的计算公式为:
测序深度 = (L × N)/ G
L:读段(reads)长度;N:读段数目;G:测序目标区域大小
覆盖度(coverage)是指测序获得的序列占整个基因组的比例。由于基因组中的高GC、重复序列等复杂结构的存在,测序最终拼接组装获得的序列往往无法覆盖所有的区域,这部分未覆盖的区域就称为Gap。值得注意的是,sequencing coverage指的就是depth of sequencing coverage也就是sequencing depth,反映了一个区域平均被多少个reads测到。
举个例子
假设对一段2000 bp(G:测序目标区域大小)的目标序列进行单端测序,得到1000条reads(N:读段数目),每条reads 200bp(L:读段长度),测序后把所有的reads比对到目标区域后,若2000bp的目标区域中有1800bp的位置至少有1个read覆盖到,而剩余的200bp没有任何read覆盖到,
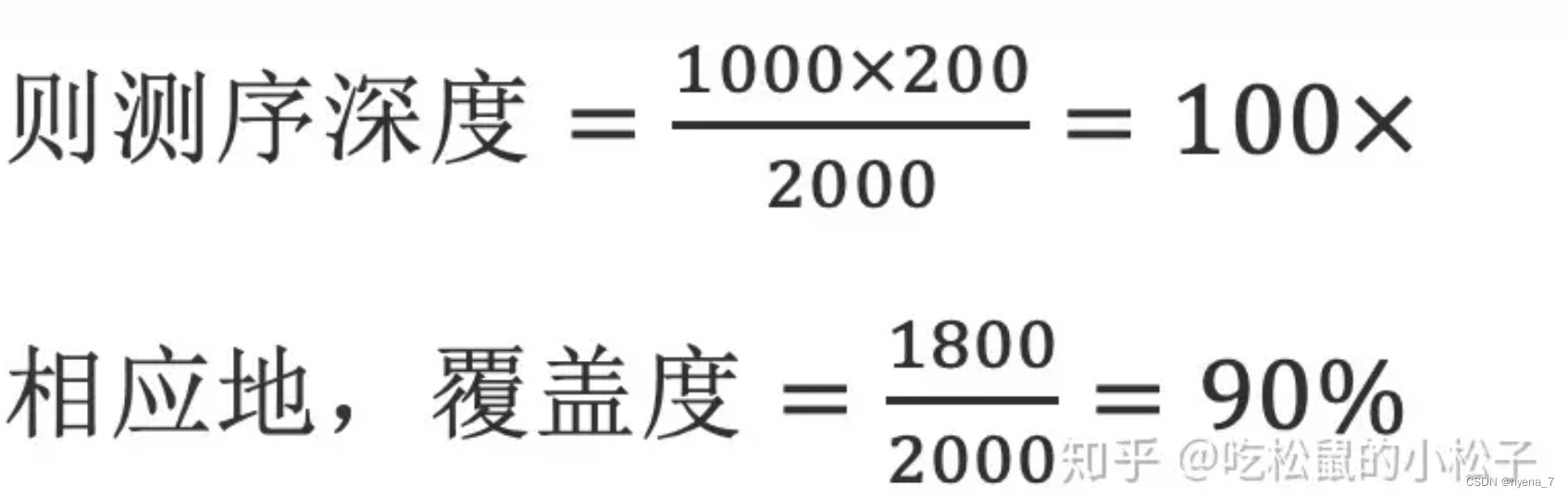
可见,测序深度与覆盖度是成正相关的。
而不同实验对于测序量的需求,与目标区域的大小、突变类型和疾病模型等是密切相关的。
对于全基因组测序(WGS)来说,人类全基因组大约3G,健康人一般需要测到30X,即获得90G有效数据;要可靠地检测基因组中的单核苷酸多态性(singlenucleotide polymorphism, SNP)和插入缺失标记(insertion-deletion, INDEL),至少需要测到35X,产生105G的有效测序数据 [1];而从头基因组测序(de novo genome sequencing)需要拼装基因组,最佳测序深度为50X [2]。
人类基因中大约有180,000个外显子,占人类基因组的1%,约30MB。对于全外显子测序(WES)来说,由于目标区域的异质性增加,以及探针50%的捕获效率,需要更大的平均读取深度才能获得与WGS相同的覆盖范围,覆盖89.6-96.8%的目标碱基,需要测到80X [1]。健康人的WES一般测到100X,获得6G数据,其中3G有效数据。
对于高异质性的细胞群或组织样本,比如肿瘤,WGS需要70-100X,WES需要160-200X。
RNA测序(RNA-seq)应用更广阔,现在已经在不同条件下的许多细胞和组织类型中进行了大量的RNA-seq实验,但是很少有关于RNA-seq测序深度的明确指南,这是因为测序要求通常取决于所研究的生物学问题,以及所测定的转录组的大小和复杂性。
单细胞真核生物和细菌等具有较简单的转录组,转录潜能也较低。如,4 M reads可以检测到80%的酵母基因。
哺乳动物拥有数以万计的基因,许多基因还包含不同亚型,并且具有不同的表达水平。在RNA-seq分析中,基因或转录本丰度通常表示为RPKM1。ENCODE2曾利用H1人胚胎干细胞做过评估,若研究对象是RPKM>10的基因,每个样本测到36 M reads就可以准确定量80%的基因表达。然而,对于低表达水平的基因(FPKM<10),要测到80 M reads才能准确定量。所以,如果需要在整个转录组准确定量所有基因(包括lncRNA基因),那么样本需要测到80M以上;如果只是研究表达量高的转录本的整体表达变化,那么每个样品36 M reads就足够了。
如果RNA-seq的目标是发现新的稀有的转录本,如noncoding RNA或mRNA新的可变剪接,考虑到这些转录本的低表达和建库方案产生的偏差,估计需要超过400 M reads。
https://zhuanlan.zhihu.com/p/74558512
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!