基因表达分析聚类&分析
1. Introduction to gene expression analysis
- Technology: microarrays vs. RNAseq. Resulting data matrices
- Supervised (Clustering) vs. unsupervised (classification) learning
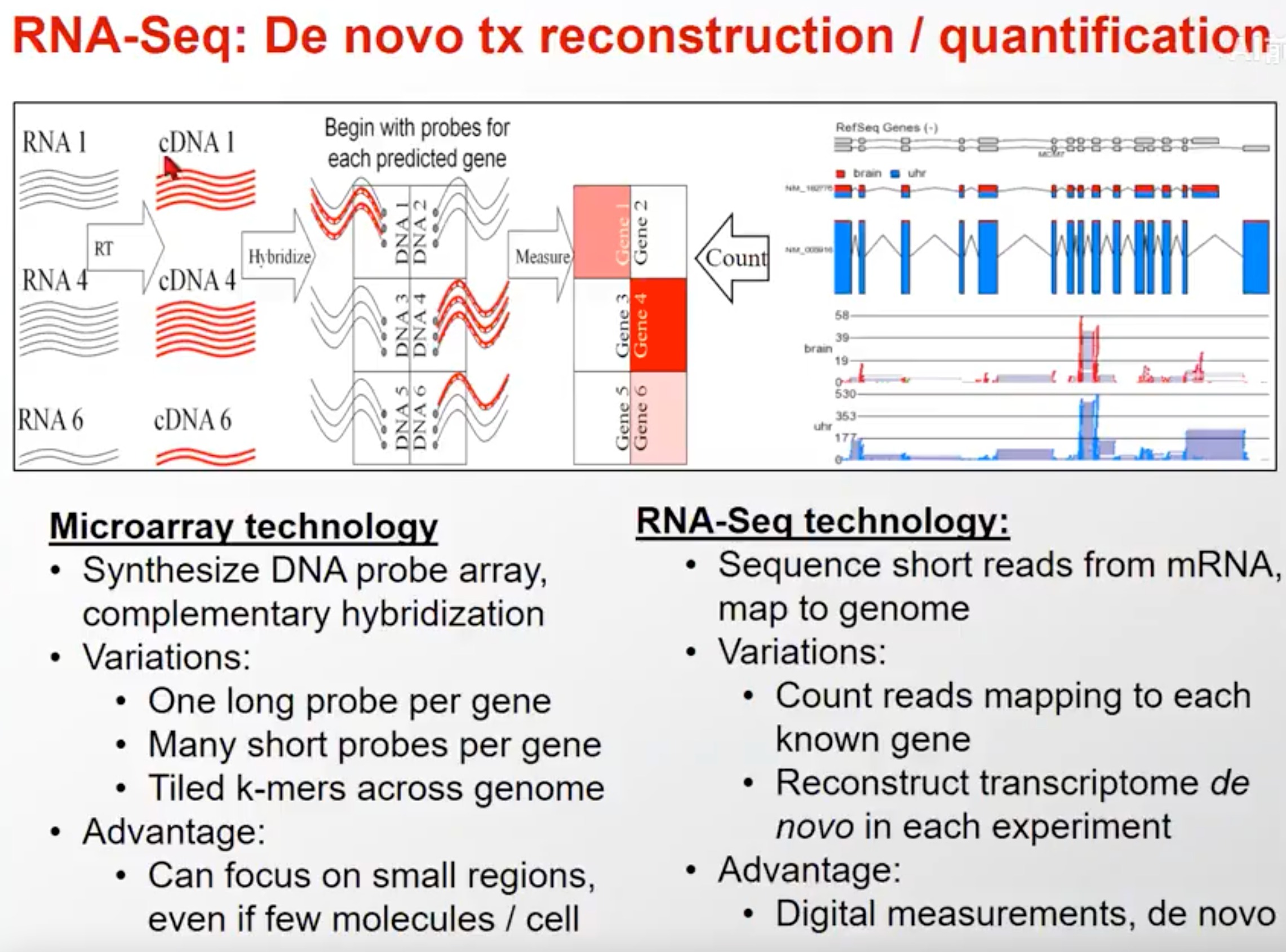
-
微阵列技术:
-
制备DNA探针阵列并进行互补性杂交。
-
变化:
- 每个基因使用一个长探针。
- 每个基因使用许多短探针。
- 在基因组中制备瓦片状k-mer阵列。
-
优点:
- 可以专注于小区域的研究,即使每个细胞的分子数目很少也可以进行。
-
-
RNA测序(RNA-Seq)技术:
-
从mRNA中测序短片段,并将其映射到基因组上。
-
变化:
- 计数映射到每个已知基因的读取数。
- 在每次实验中重建转录组(reconstruct transcriptome de novo)。
-
优点:
- 提供数字化的测量,每次实验都可以进行新的转录组重建(de novo transcriptome reconstruction)。
-
微阵列技术:是一种生物技术,它允许研究者在单一实验中测量数千甚至数万基因的表达。这是通过创建包含许多特定DNA探针的阵列来实现的。这些探针可以设计为特异性地结合到特定的mRNA目标,从而允许研究者定量地测量每个基因的mRNA表达。微阵列技术可以提供全基因组的表达画像,帮助研究者了解不同条件下基因表达的变化。
RNA测序(RNA-Seq)技术:是一种基因表达剖析的先进技术。它使用下一代测序(NGS)技术,**从mRNA生成一系列短片段或“读取”,然后这些读取可以映射回参考基因组,从而确定它们来自哪个基因。**RNA-Seq技术比微阵列更精确,能检测更广泛的基因表达水平,也能发现新的转录和剪接事件。
值得注意的是,这两种技术各有优缺点。例如,微阵列技术成本较低,但其检测范围有限,只能测量已知的基因。而RNA-Seq虽然能提供更详细的信息,但成本更高,数据处理也更复杂。
DNA探针的制备一般涉及以下步骤:
- 确定目标:选择需要检测或分析的特定DNA序列作为目标。
- 合成探针:通过生物化学方法,合成一段与目标序列完全互补的DNA片段,即DNA探针。
- 标记:将探针与放射性、荧光或酶等标记物结合,以便在后续实验中检测和跟踪。
- 杂交:将标记的探针与待测样品进行杂交,探针会寻找并结合到与其序列完全匹配的DNA或RNA片段。
- 检测:通过放射性、荧光或酶等方式检测探针,从而确定目标序列是否存在,以及存在的数量。
基因测序之后就可以得到基因表达阵列,并可以对其进行分析
每一行代表不同的基因,每一列对应着不同的实验条件(如不同的组织细胞/不同疾病/不同人群/以及等等其他)
- 对于每行数据,你可以看到同一个基因在不同实验下的差异表达
- 对于每列数据,你可以看到不同试验下,整条基因组的差异
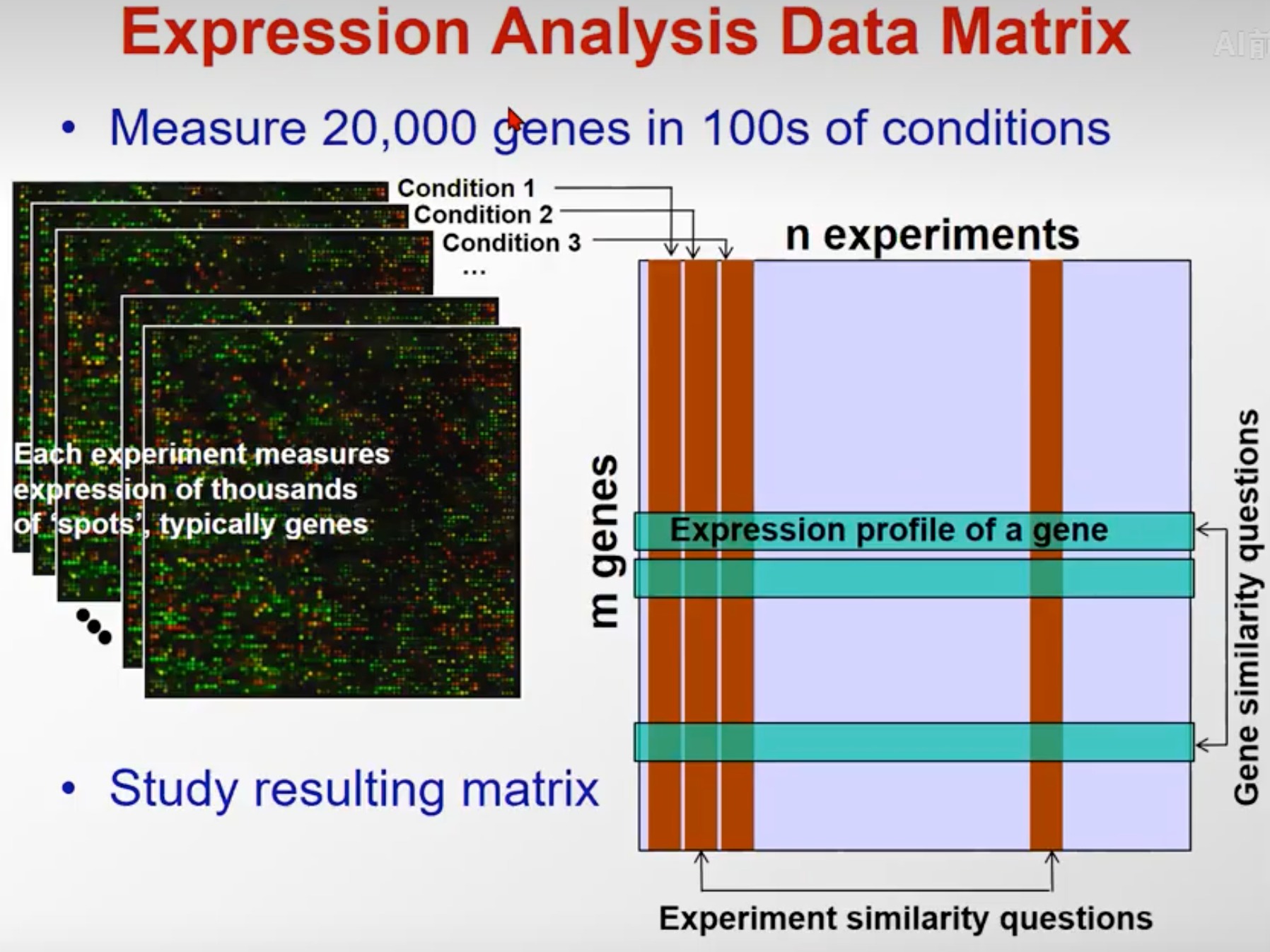
- 对于基因测序数据,存在着很多潜在的任务,最典型的就是 聚类和分类 两种问题
- Clustering【无监督学习】: 具有相似功能的基因表达往往会富集在一处,因此也揭示了潜在的结构
- 注释:特定基因功能
- 不提前注释
- Classification【监督学习】: 从数据中提取特征,并且强制机器去学到这些不同类别之间数据特征的差异
- 提前有注释,分类好的集群
- Clustering【无监督学习】: 具有相似功能的基因表达往往会富集在一处,因此也揭示了潜在的结构

评估聚类性能

聚类是一种无监督学习方法,常用于探索数据集中的自然分组或模式。在生物信息学中,聚类常常被用于基因表达数据分析,以发现具有相似表达模式的基因群。
这里提到了两种常见的评估方法:鲁棒性和类别富集。
-
鲁棒性: 这种方法主要用于评估聚类的稳定性。这通常涉及到从数据集中随机选择样本进行聚类,然后重复这个过程。如果某个聚类在所有的子样本中都出现,那么我们就可以认为这个聚类是鲁棒的。如果聚类结果在多次随机抽样后变化不大,那么我们就可以认为这个聚类方法是稳定的。
-
类别富集:这种方法主要用于寻找在特定聚类中“过度表达”的基因类别。这是一种后验验证方法,可以用于评估聚类结果的生物学意义。例如,如果一个聚类包含了许多在某种生物过程中起作用的基因,那么我们就可以认为这个聚类可能与这个生物过程有关。这种方法也常常用于基因表达模式的发现。
- 使用超几何分布来评估聚类结果
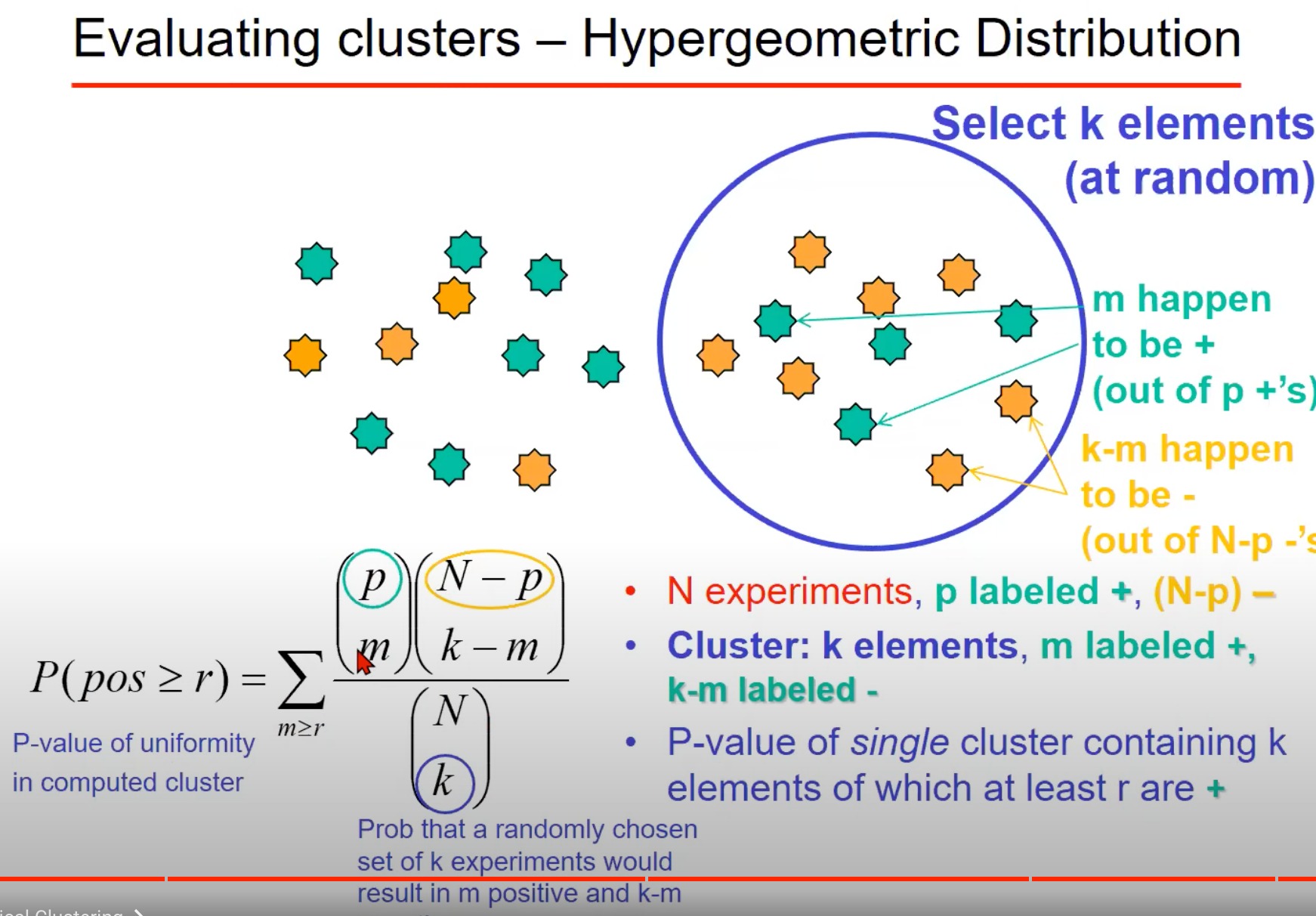
超几何分布是一种描述从有限的两类对象(例如正例和负例)的总体中无放回地抽取的概率模型。在生物信息学中,这种方法经常用于基因富集分析,以评估某个基因集合(例如一个聚类)中特定基因类别的富集情况是否超过了随机期望。
-
为了具体说明,我们设
- N为总体的大小
- p为总体中标记为"+"的元素数量
- N-p为标记为"-"的元素数量。
- 我们随机抽取k个元素,其中m个标记为"+“,k-m个标记为”-"。
-
超几何分布可以帮助我们计算出,在这种情况下,随机抽取k个元素中至少有r个"+"的概率。这个概率可以用来评估我们观察到的聚类中特定类别的基因数量是否比随机期望的要多。
-
"P-value of uniformity"和"P-value of single cluster containing k elements of which at least r are +"就是根据超几何分布计算出来的p值。
- 如果p值很小(例如小于0.05),那么我们就可以认为观察到的富集情况是非常罕见的。
- 因此可能不是随机产生的,而是有一些生物学的原因。
- 这样就可以帮助我们找出聚类结果中生物学上有意义的模式。
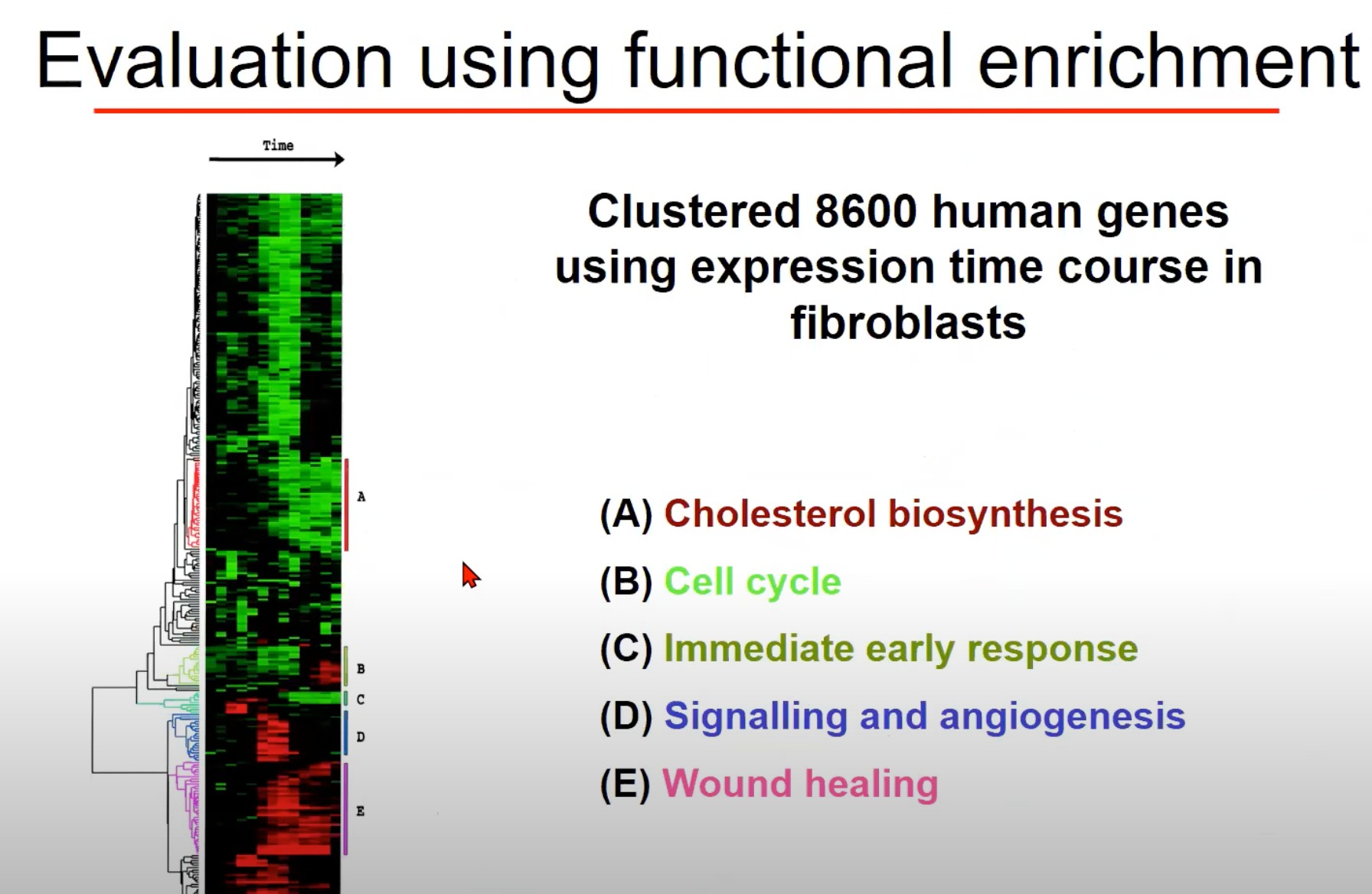
通过使用人类基因在成纤维细胞中的表达时间序列进行聚类分析。这个聚类分析将8600个人类基因划分到了五个主要的类别中。
A) 胆固醇生物合成:这个类别的基因可能主要涉及在细胞中生产胆固醇的过程。
B) 细胞周期:这个类别的基因可能主要参与控制细胞的生命周期,包括细胞的生长、DNA复制、分裂等过程。
C) 即时早期反应:这个类别的基因可能主要涉及细胞对各种刺激的快速反应,例如应对环境变化、压力或损伤。
D) 信号传导和血管生成:这个类别的基因可能主要参与细胞间的通信,以及血管的形成和发展。
E) 伤口愈合:这个类别的基因可能主要参与伤口修复和再生的过程。
每个类别都是由在相似条件下表达的基因组成的。这意味着这些基因可能在相同的生物过程中起作用,或者受到相同的调控机制影响。这种类型的分析对于理解基因的功能,以及它们如何在复杂的生物过程中协同作用非常有帮助。
总结

- 两种分类方法
在分类问题中,通常有两种主要的方法:生成方法(Generative)和判别方法(Discriminative)。
-
生成方法(Generative):
- 生成方法试图学习数据的联合概率分布P(X, Y),然后使用贝叶斯定理来推导出条件概率分布P(Y|X)。生成模型能够产生新的数据样本,这是它们名字的由来。代表性的算法有贝叶斯分类器(例如朴素贝叶斯)和隐藏马尔可夫模型(HMM)等。
- 生成模型将分类问题描述为概率问题,它在不同类别中建模特征分布,并使用概率计算进行决策。在基因发现(Gene Finding)问题中,隐藏马尔可夫模型就是一种常用的生成模型。
-
判别方法(Discriminative):
- 与生成方法不同,判别方法直接学习决策边界或者条件概率分布P(Y|X)。判别模型并不对数据的分布做假设,它们直接学习输入和输出之间的映射关系。代表性的算法有支持向量机(SVM)、决策树、随机森林、逻辑回归、深度学习等。
- 判别模型不对底层分布进行建模,而是使用距离边界的距离来进行决策。在基因发现问题中,条件随机场(CRF)就是一种常用的判别模型。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- windows文件名命名规范(文件名规范、命名规则、避免特殊字符、注意文件名长度限制260个字符)
- 【教3妹学编程-算法题】找到 Alice 和 Bob 可以相遇的建筑
- echarts tooltip 提示框阴影设置
- STL容器之string(上)
- 类与对象(上)
- word2019保存后的图片变模糊了怎么办?Word 2019 默认保存后压缩变模糊的问题,解决方案
- 数据库:园林题库软件(《城市绿地设计规范》答题卷二 )
- 极智芯 | 解读自动驾驶芯片之寒武纪行歌SD系列
- 第三章 java内存模型与并发三大特性
- 玩转Mock.js:构建模拟数据的利器