5+共病+WGCNA+实验。共病+实验搭配是经典的得分套路
今天给同学们分享一篇生信文章“Adipocyte dysfunction promotes lung inflammation and aberrant repair: a potential target of COPD”,这篇文章发表在Front Endocrinol (Lausanne)期刊上,影响因子为5.2。

结果解读:
肥胖对慢性阻塞性肺疾病的影响在不同的临床研究中存在不一致的结果
在表1中列出了15项关于COPD和肥胖的研究。其中4项研究报告称超重或肥胖对COPD患者的生存有益,而3项研究显示超重或肥胖的患者患急性加重的风险降低。然而,另外3项研究报告称肥胖可能会影响COPD患者的运动能力。一些大规模的横断面研究表明,超重或肥胖的人患COPD的发病率增加,这可能与对香烟烟雾或污染物的反应加剧有关。因此,目前的研究对超重或肥胖对COPD的影响存在不一致的情况。
在COPD和肥胖数据集中发现了差异表达基因(DEGs)
为了探索基因表达的固有模式和COPD与肥胖之间的潜在关联,作者分析了GEO公共数据集中的DEGs。在过滤重复和无意义基因后,共鉴定出342个COPD相关DEGs和223个肥胖相关DEGs,其中包括18个交集的DEGs(图1A)。DEGs的火山图绘制在附图1A和附图1B中。炎症因子[C-X-C Motif Chemokine Ligand 8 (CXCL8)、CXCL2和基质金属蛋白酶9 (MMP9)]在肥胖患者中显著增加,而脂质代谢调节因子(Apolipoprotein B (APOB)和Cholesteryl ester transfer protein (CETP))在肥胖患者中显著降低。对于COPD患者,BMPR2通路的关键调节因子Activin A Receptor Type 2B (ACVR2B)和Transforming Growth Factor Beta 1 (TGFβ1)显著下调,与肥胖相关的基因Apelin Receptor Early Endogenous Ligand (APELA)也显著下调,然而BMP4显著上调。肥胖和COPD中鉴定的18个交集DEGs详见附表2、3。在这些基因中,作者发现蛋白激酶Cβ(PRKCB)的表达水平在肥胖症中较高,而在慢性阻塞性肺疾病(COPD)中较低。与对照组相比,磷脂酰肌醇-3-激酶调节亚单位2(PIK3R2)和还原型烟酰胺腺嘌呤二核苷酸磷酸酶1(NOQ1)基因在肥胖症和COPD中分别增加(图1B)。
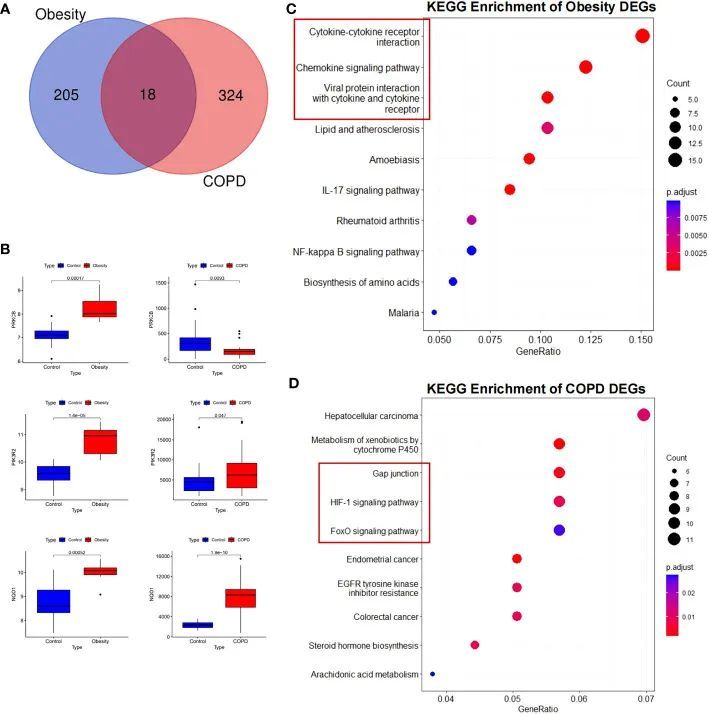
KEGG、GSEA 和 PPI 分析强调了调节炎症和损伤修复的脂肪细胞因子
KEGG对肥胖症中的差异表达基因(DEGs)的注释分析显示,该途径主要包括细胞因子-细胞因子受体相互作用(P = 4.17E-05)、病毒蛋白与细胞因子及细胞因子受体的相互作用(P = 9.32E-06)、趋化因子信号通路(P = 4.17E-05)以及脂质和动脉粥样硬化(P = 0.0029)(图1C)。至于COPD,DEGs的KEGG途径分析显示明显富集于缝隙连接(P = 0.003734)、细胞色素P450代谢异物(P = 0.002103)、缺氧诱导因子1-alpha(HIF-1)信号通路(P = 0.008497)以及Forkhead box O(FoxO)信号通路(P = 0.02568)(图1D)。同样,通过GSEA,作者确认细胞因子和细胞因子受体基因在COPD和肥胖症中同时上调,但在这两种病理状态中呈相反的趋势(附图1C、D)。
为了确定上述差异表达基因(DEGs)之间的相互作用,作者还通过STRING分析生成了蛋白质相互作用网络(PPI网络)。发现CXCL8、MMP9和C-C Motif趋化因子受体5(CCR5)等脂肪细胞因子和蛋白酶是肥胖患者白色脂肪组织中的关键基因。此外,细胞连接蛋白和生长因子,如Catenin beta-1(CTNNB1)、表皮生长因子(EGF)、表皮生长因子受体(EGFR)和脑源性神经营养因子(BDNF),是COPD患者气道上皮细胞中的关键基因。作者的结果表明,炎症和损伤修复是肥胖和COPD中的主要共同特征。
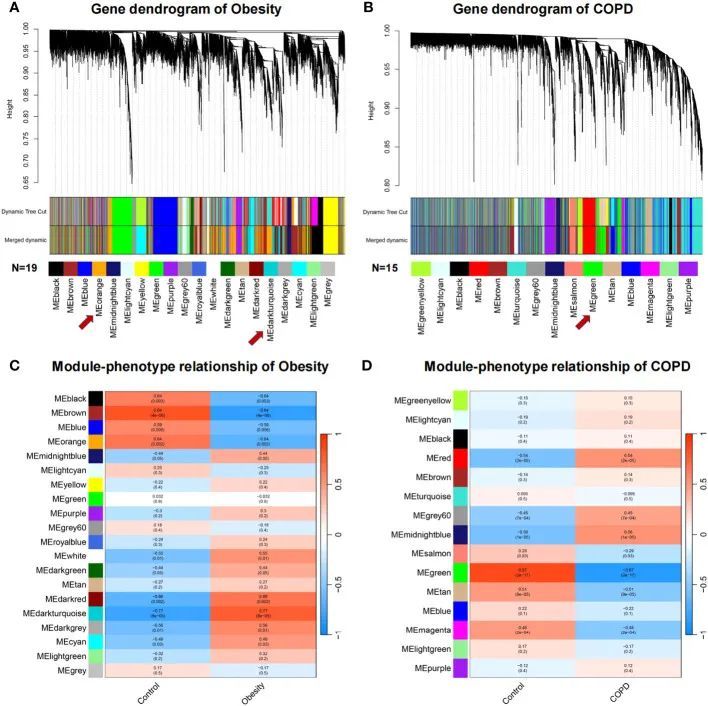
WGCNA 分别揭示了肥胖和COPD 的关键模块
为了描述基因的共表达关系,分别对与肥胖和COPD密切相关的高度相关基因模块进行了WGCNA分析。共表达模块通过聚类树状图展示。肥胖组和对照组显示了19个基因模块,如图2A中的叶子和分支所示。MEbrown与肥胖最显著相关(相关值为0.84;P = 4E-06)。差异表达基因(DEGs)和关键基因主要分布在MEorange(相关值为-0.64;P = 0.002)和MEdarkturquoise(相关值为0.77;P = 8E-05)模块中,而BMPR2分布在MEmidnightblue模块中(相关值为0.44;P = 0.05)(图2C)。在COPD和对照组中,有15个基因模块可以富集(图2B),其中MEtan显示了最显著的相关性(相关值为-0.51;P = 9E-05)。差异表达基因(DEGs)和关键基因主要分布在MEgreen(相关值为-0.87;P = 2E-17)中,而BMPR2位于MEturquoise(相关值为0.095;P = 0.5)中(图2D)。
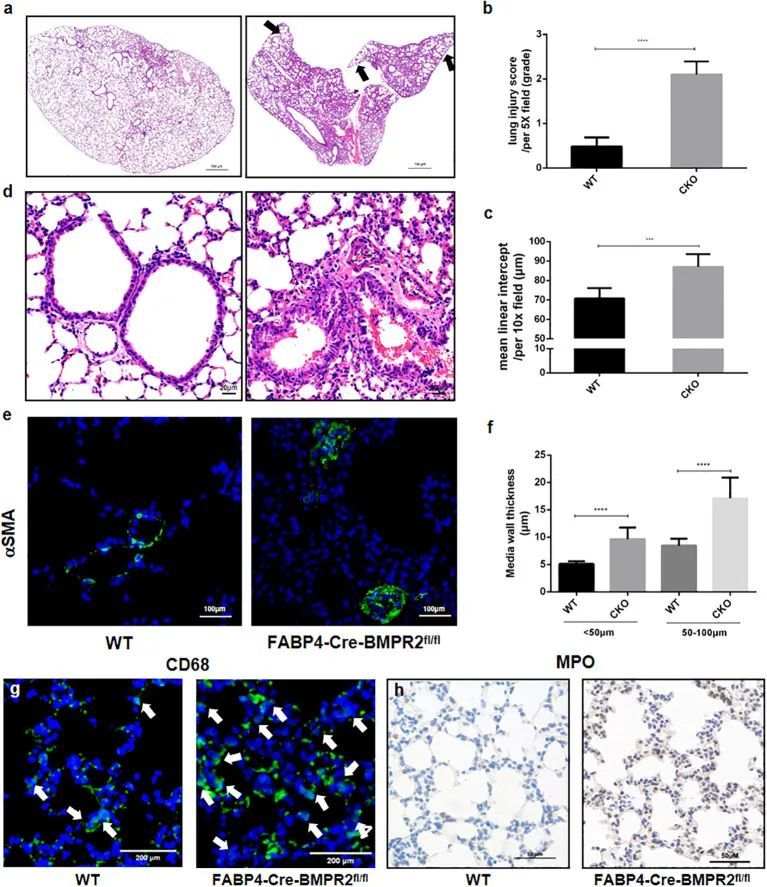
脂肪细胞特异性BMPR2基因敲除导致肺损伤和肺气肿
BMPR2在维持肺血管稳态和PAH的发展中起着关键作用,而BMPs是其配体。作为肥胖和COPD中18个交集DEGs之一,PRKCB与BMPR2有PPI。SPP1也与BMPs有相互作用。此外,从肥胖和COPD的PPI网络结果来看,COPD-PPI中的关键基因与BMPR2有密切关系。因此,BMPR2可能在与COPD相关的肺损伤和肺气肿中发挥重要作用。
作者证明了FABP4-Cre-BMPR2 fl/fl (CKO)小鼠表现出自发性肺炎和明显增加的肺损伤评分,与WT小鼠相比(图3A,B)。CKO小鼠还出现了外周肺气肿,肺外周的平均线性间隔增加(图3A,C)。此外,图3D-F显示小血管壁适度增厚,肺动脉平滑肌细胞(PASMCs)肥大。至于炎症细胞,作者观察到CD68标记的巨噬细胞和MPO标记的中性粒细胞在CKO小鼠的肺泡中明显上调(图3G,H)。有趣的是,肺炎、肺气肿和血管重塑也是慢性阻塞性肺疾病的病理特征。作者假设加重的炎症和小动脉重塑共同导致右心功能障碍,然而3周以下的小鼠缺乏成熟的代偿机制,容易死亡。
为了进一步研究机制,作者比较了野生型(WT)组和CKO组之间肺细胞增殖和凋亡的差异,通过对肺切片进行免疫染色(增殖细胞核抗原-PCNA)和TUNEL染色。PCNA主要在气道中表达,在两组中没有差异(图4A)。CKO组中观察到轻微增加的凋亡细胞数量(图4B)。有趣的是,BMPR2及其靶向蛋白Id2在CKO小动脉中下降,但在小气道中没有下降(图4C,D)。这表明脂肪组织中BMPR2的缺乏可能会影响其在其他器官中的表达,尤其是在肺血管中。此外,收缩素前体内皮素在CKO小气道和小动脉中上调(图4E),但内皮素转化酶1在两组中相似。与炎症细胞一致,CKO小鼠肺泡中的MMP2也增加(图4F)。

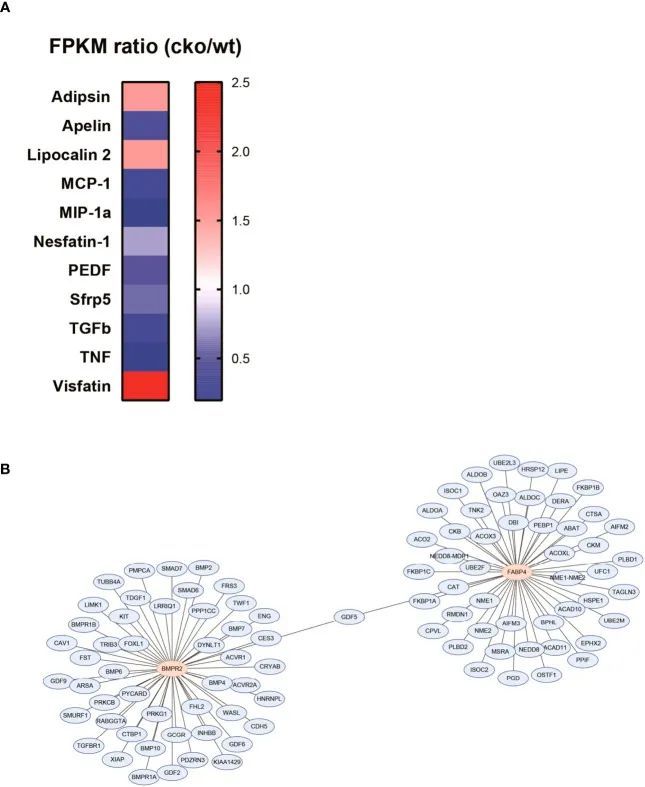
脂肪细胞因子参与了BMPR2缺乏的脂肪细胞引起的肺损伤的发病机制
为了初步探索BMPR2缺乏的脂肪细胞的作用,作者重新分析了WT和CKO小鼠脂肪细胞的RNA序列数据。每组脂肪细胞来自四只个体小鼠的腹股沟脂肪组织。作者比较了多种脂肪细胞因子的差异表达,并列出了具有外显子模型每百万映射片段的碱基对数大于1和折叠变化大于1.5的脂肪细胞因子(图5A)。作者观察到CKO组中apelin显著减少,adipsin略有增加(图5A)。此外,作者搜索了与BMPR2和FABP4相关的前50个蛋白质,并确定了唯一的共同基因(图5B)。因此,作者推测apelin、adipsin和生长分化因子5(GDF5)可能参与了BMPR2缺乏的脂肪细胞影响肺结构的病理过程。
总结
总之,炎症和异常修复可能是肥胖与COPD之间病理关联的潜在机制。此外,这项研究创新地补充了脂肪细胞功能障碍与肺损伤之间的病因联系,表明脂肪细胞可能成为COPD发展中的一类关键细胞。这为COPD的病理机制和有希望的治疗提供了新的见解。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- split(“ “) 和 split(“\s+“) 的区别
- Vue中转换HTML为PDF
- DAY10--learning english
- 【局域网window10系统搭建共享文件夹或与手机共享】
- 第四章 配置本地安全策略
- ESP8266 ESP-01/01s 工作模式与固件下载烧录接线
- 项目经理周报,月报编写模板
- Python模块与包
- Baumer工业相机堡盟工业相机如何通过NEOAPI SDK实现相机掉线自动重连(C++)
- 实现大数相减,完整版