【Gene Expression Prediction】Part2 Enchancer discovery
文章目录
来自Manolis Kellis教授(MIT计算生物学主任)的课
YouTube:(Gene Expression Prediction - Lecture 09 - Deep Learning in Life Sciences (Spring 2021)
Slides: slides
本节课分为四个部分,本篇笔记是第二部分。
本节主要是一个讲座STARR-seq,探讨发现增强子的方法。如何利用弱监督学习检测增强子。评估了模型性能,以及它们在基因表达分析中的应用潜力。
5. 第一个讲座:Enchancer discovery


增强子是增强特定基因的转录的。对理解细胞如何在不同条件下调控基因表达至关重要。
以前的识别方法都是一些非监督方法,因为没有监督数据
5.1 STARR-seq
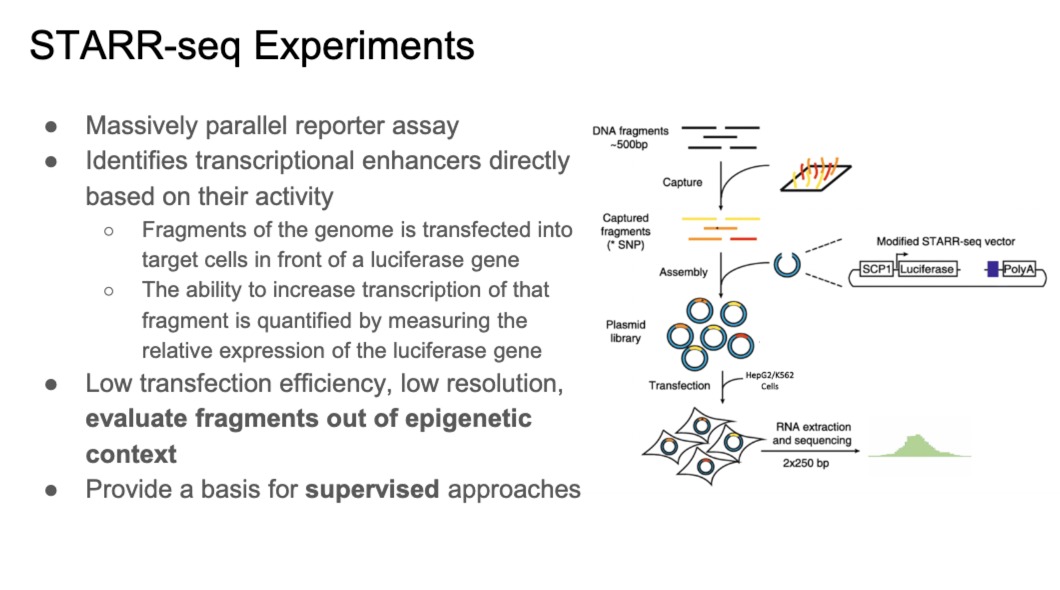
MPRA(Massively Parallel Reporter Assays)是一种高通量基因组功能验证技术。它可以同时测试成千上万个DNA序列(如启动子、增强子等调控元件)对基因表达的影响。
大量候选DNA序列插进短的报告基因vector中(比如荧光素酶基因,都包含一个小的启动子)。如果一个候选DNA是增强子,就会增强基因转录表达,就会被检测出来。
关键优点是能够在一个实验中评估大量序列,这是传统单个报告基因测定所无法实现的
STARR-seq 是MPRA的一个变体。
在一个细胞系中,它通过直接测量基因片段的活性来识别转录增强子。
产生数据为监督学习提供基础,DNA序列和其活性标签。
这里主要是为了大规模筛选,所以就用了统一的报告基因,就是为了测试DNA序列的增强子活性

不同细胞类型(如K562、HepG2等)进行的一系列基因组分析实验。
ENCODE项目研究人员的一个核心假设,即染色质的开放状态和组蛋白修饰之间的相互作用是调控基因表达特别是转录因子结合和增强子活性的关键。
这些数据集用于进行验证
在这里就是我们能否使用这些特征,来验证STARR-seq expression
5.2 Enchancer detection with weakly supervised learning
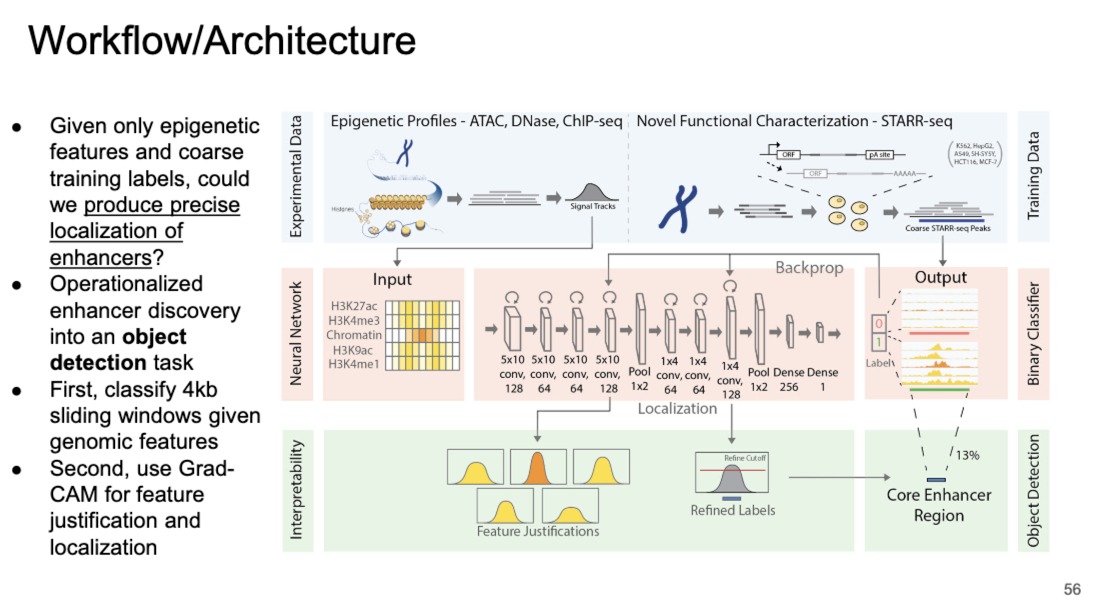
该模型结合了表观遗传特征数据(如ATAC-seq、DNase-seq和ChIP-seq的信号轨迹)和STARR-seq实验结果来训练一个模型,从而识别和精确定位增强子
-
首先模型的input,就是某个区域的,4kb大小的滑动窗口,进行移动
- 记录每个窗口的表观遗传标记的数据,包括组蛋白修饰的分布、DNA可及性的测量等等。(基因组上每个位置的综合信号强度)
- 按我的理解的话,就是一个input matrix就是4kb的窗口
-
然后output是STARR-seq实验得到的数据。
- 包括增强子位置区域预测和核心增强子区域(真正起到增强作用的)
-
神经网络使用这些数据来预测哪些窗口包含活跃的增强子区域。
-
Grad-CAM技术可以帮助分析决策过程,有助于科学家们理解模型为什么会认为某个特定区域是一个增强子。
-
这里其实是重点的一个部分
-
使用在训练过程中计算出的梯度信息,这些信息表示了模型输出(如增强子的预测位置)对输入特征的敏感性
-
可以看出哪些地方对决策贡献更大
-
例如,如果Grad-CAM揭示了在某个4kb窗口内,H3K27ac的特定模式与模型预测增强子存在密切相关,那么我们可以得出结论,这种特定的组蛋白修饰模式对于识别增强子是重要的。
-
这样的准则是更加通用的
-
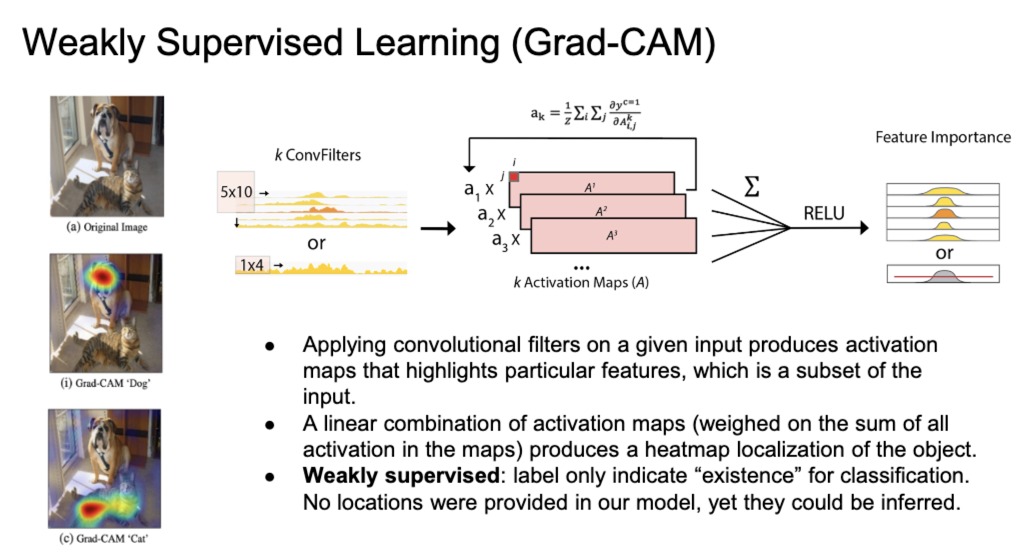
如下图所示,Grad-CAM在弱监督学习中应用效果好。弱监督学习就比如只能告诉我们照片里有猫/狗,但是我们不知道具体位置
跟我们这里只知道增强子活动而不知其位置的道理是一样的
-
卷积层中的激活图
- 每个卷积核都会产还是呢个一个激活图,高亮了图像中相关特征的位置
-
梯度相加
- 对于所选的类别(如“狗”或“猫”),网络在最后一个卷积层的每个激活图上计算该类别相对于该激活图的梯度。
- 这些梯度表示激活图对于识别该类别的重要性。
- 梯度越大,越关键
-
组合生成热图
- 将这些梯度加权的激活图进行线性组合,得到最终的Grad-CAM热图
- 通过ReLU函数处理,保留对类别有正贡献的特征,取消无关特征。
-
在基因组学中
- 通过Grad-CAM,可以理解模型在预测基因组上的特定区域为增强子时,哪些表观遗传标记起了决定性作用。这有助于解释模型的决策过程,并可以指导进一步的生物实验来验证这些预测。
5.3 Model performance

一个基因组学模型在不同细胞系和染色体上的交叉验证
- 细胞系交叉验证:
- 模型使用一个或多个细胞系的数据进行训练,并在其他未使用的细胞系数据上进行测试。
- 泛化到新的细胞系的能力。
- 染色体交叉验证(Leave-One-Chromosome-Out Cross Validation):
- 模型在除了一个染色体的所有染色体数据上进行训练,并在剩下的那个染色体上进行测试。
- 验证模型是否能够泛化到基因组上新的位置。

左边的是预测出来增强子在的区域,并且给出了表观遗传标志和基因注释、预测标签
然后论文中该模型性能超过SOTA(这里不展示)

举例一下,关于神经祖细胞(NPC)的案例研究。
-
Grad-CAM技术分析
- 左图:不同表观遗传标记在4kb基因组窗口内的重要性评分。预测贡献。
- 右图:展示了窗口中每个位置的重要性评分。描绘出潜在增强子的精确位置。(就像检测猫在图中的哪个位置一样)
-
预测统计
- 总覆盖面积(Total Coverage):经过精细化的预测覆盖了更小的区域
- TSS重叠(TSS overlap):显示精细化后的预测与TSS的重叠比例显著增加(从原始的数值增加到71.0%),表明预测与已知的基因表达起始区域更加吻合。
- PhastCons得分:PhastCons是一种评估跨物种保守性的方法,得分增加(p-value < 0.001)表明经过精细化的预测区域在进化上更加保守,可能具有重要的生物学功能。
- 稀有衍生等位基因频率(Rare Derived-Allele-Frequency, DAF)SNP富集:稀有DAF SNP的富集表明这些区域在同种内也是受到选择压力的,进一步暗示预测的增强子在功能上是重要的。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- MOSFET管驱动设计细节,波形分析
- 【Helm 及 Chart 快速入门】03、Chart 基本介绍
- 安科瑞AEM96系列三相多功能碳结算电能表——安科瑞赵嘉敏
- kafka之java客户端实战
- linux下安装JRE
- QT文件介绍
- 分布式定时任务系列8:XXL-job源码分析之远程调用
- C++播放音乐:使用EGE图形库
- 提升思维能力,高效管理信息——推荐使用SimpleMind Pro(思维导图)应用
- freeRTOS总结(四)中断管理