Seer*Stat乳腺癌数据预测模型-Step1数据清洗和预处理
发布时间:2024年01月14日
通过R语言进行数据的清洗
由于刚从Seer上面下载的数据需要进行预处理,不然模型是无法读懂字符串的,也有一些数据的预处理操作也要执行。
进入R官网下载环境
https://www.r-project.org/ 这是官网,进入官网之后,点击download R即可
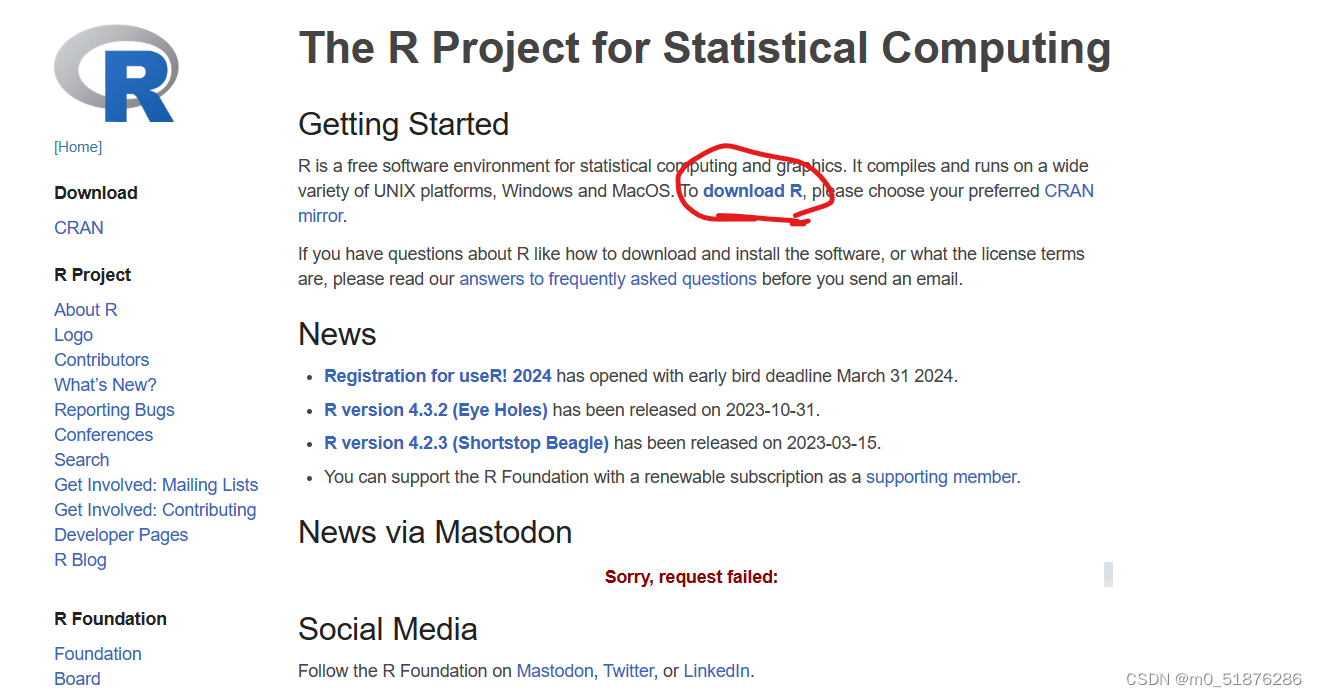
随意选择一个镜像源进行下载即可,这里我选择的是清华大学的镜像源

这里根据自己的系统来进行下载即可
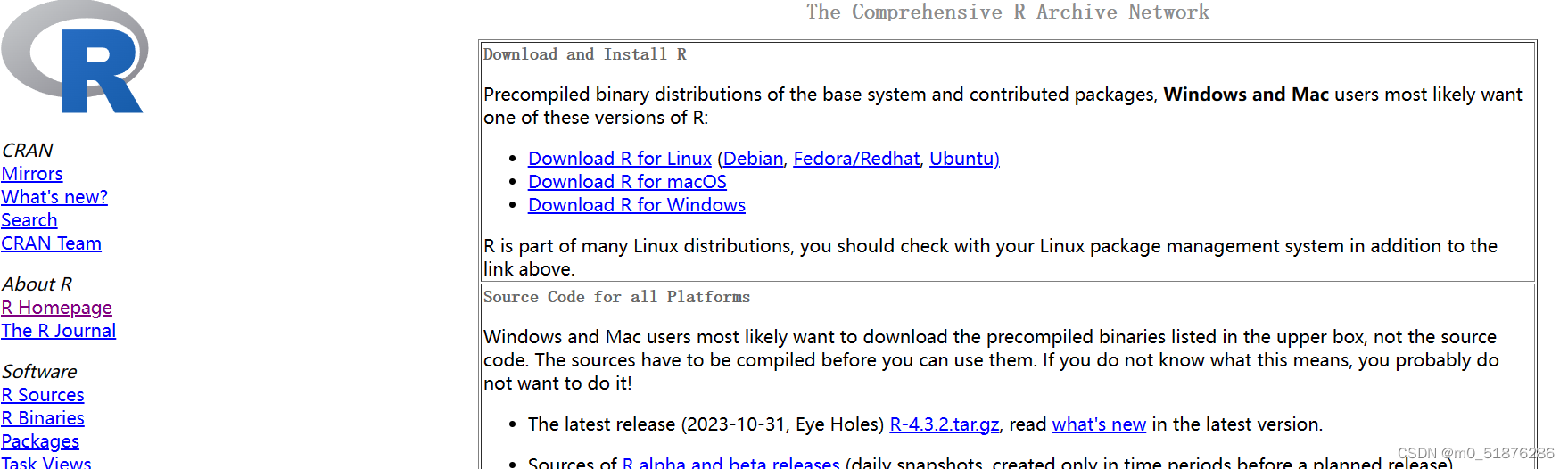
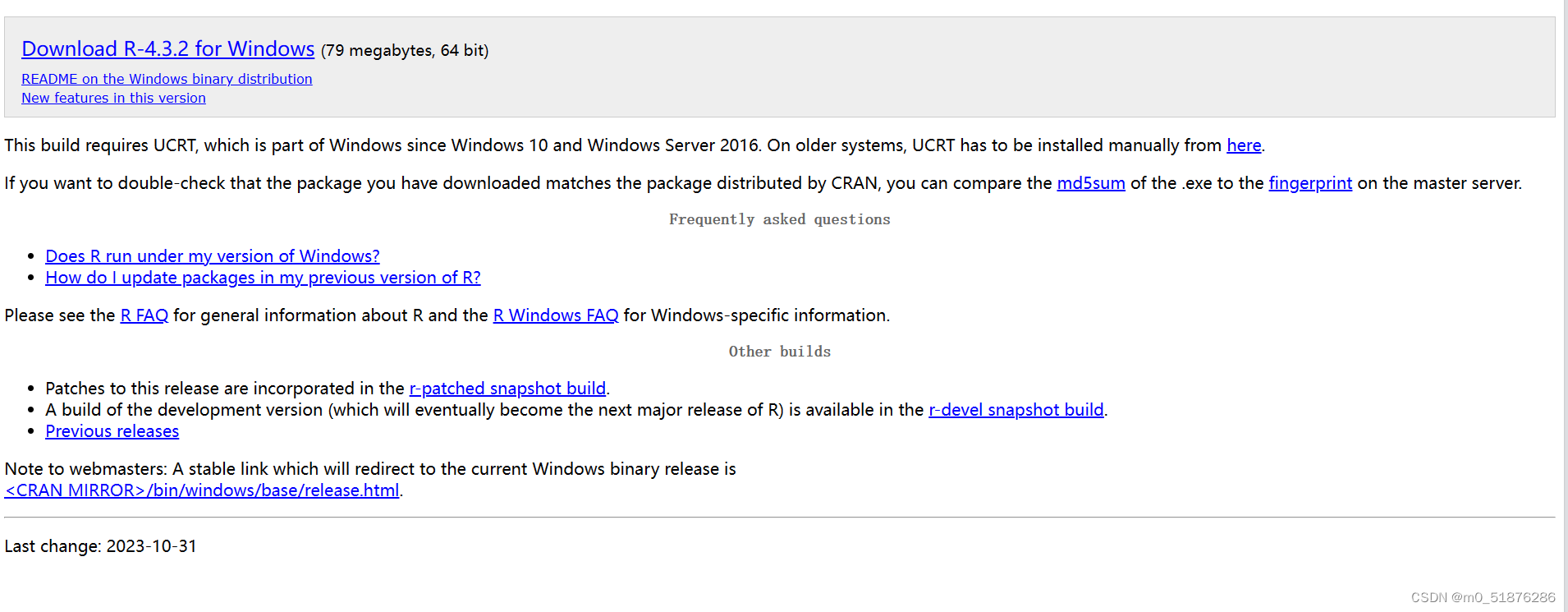
进去之后下载base 和Rtools
base
Rtools
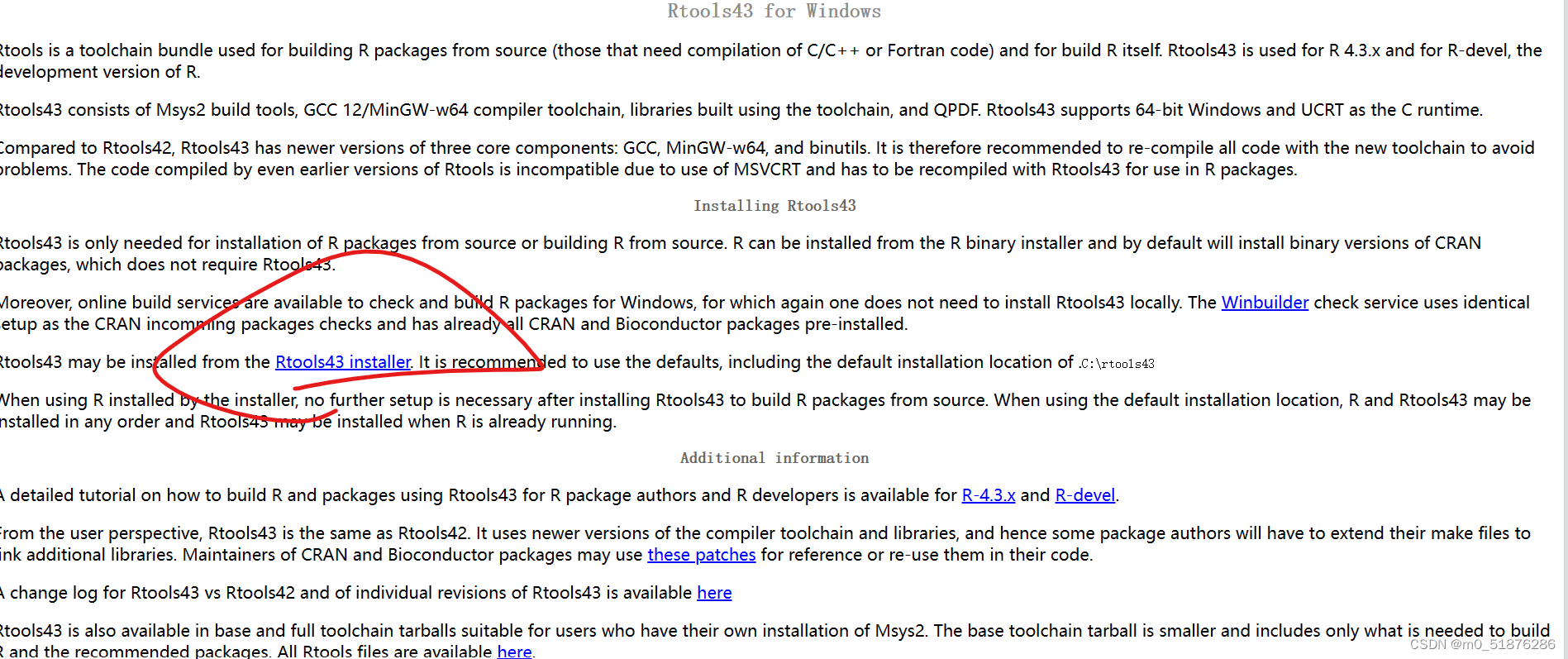
下载安装程序之后记住安装路径不要出现中文路径就行,要不然会有很大的问题。其中建议Rtools的安装路径不要修改就默认。
下载IDE:Rstudio
https://posit.co/downloads/ 这个是官网
也可以直接从这个官网下载R环境也行。
安装好之后打开即可
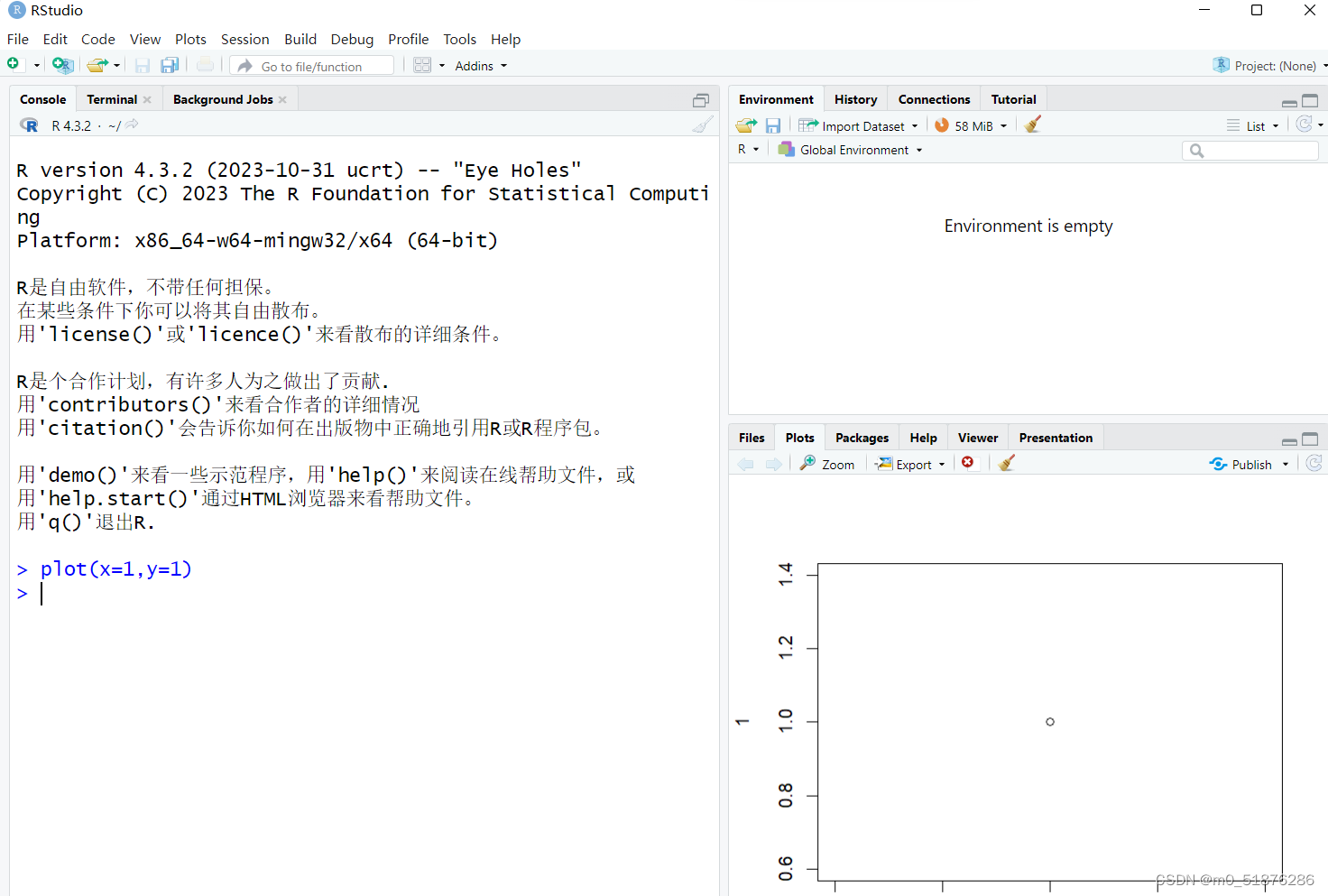
因为R包的下载一般都是在外网下载有可能不成功,所以要换成国内的镜像,配置文件就是在你安装的路径下面,etc目录里面插入以下代码
## 设置镜像
local({r <- getOption("repos")
r["CRAN"] <- "https://mirrors.tuna.tsinghua.edu.cn/CRAN/"
options(repos=r)}
)
options(BioC_mirror="https://mirrors.ustc.edu.cn/bioc/")
## 设置下载方式
options("download.file.method"="libcurl")
options("url.method"="libcurl")
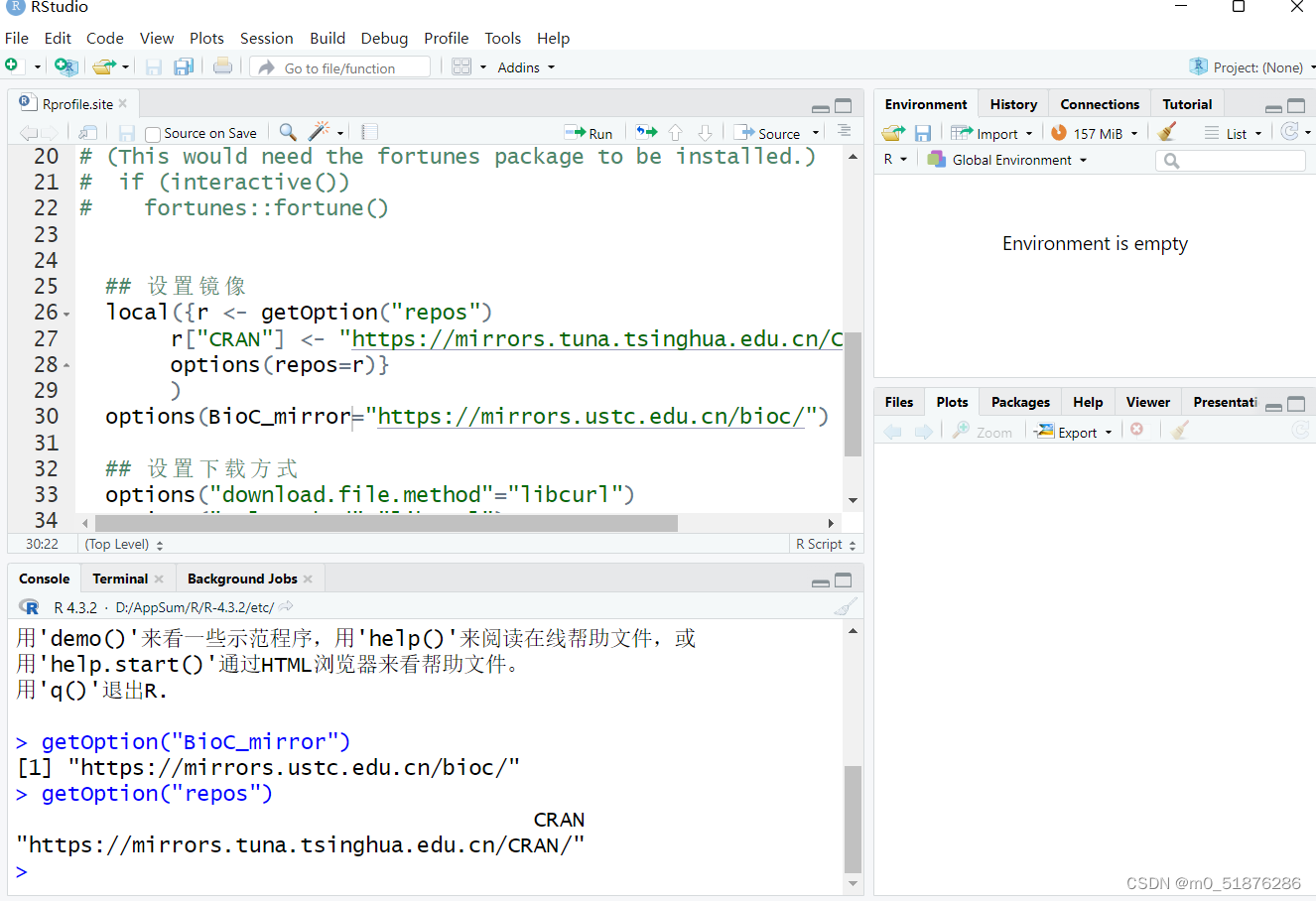
验证是否修改完成,保存之后再次打开通过下面的方式进行验证
一些基本的小命令
.libPaths():查看R包安装的位置installed.packages():查看已经安装好的包available.packages():查看可安装的R包library(包名):查看某个包是否存在
安装CARN上的包(一些比较常用的包)
update.packages()
if(!require("xlsx")) install.packages("xlsx")
if(!require("tidyr")) install.packages("tidyr")
if(!require("dplyr")) install.packages("dplyr")
if(!require("ggplot2")) install.packages("ggplot2")
if(!require("data.table")) install.packages("data.table")
if(!require("ggrepel")) install.packages("ggrepel")
if(!require("devtools")) install.packages("devtools")
if(!require("BiocManager")) install.packages("BiocManager")
安装Bioconductor上的包
BiocManager::install()
if(!require("DESeq2")) BiocManager::install("DESeq2")
if(!require("clusterProfiler")) BiocManager::install("clusterProfiler")
使用Rstudio对Seer乳腺癌数据进行清洗
前提工作:需要下载foreign,car,stringrs三个R包,如果没有就下载即可。
将下载的数据读入Rstudio中
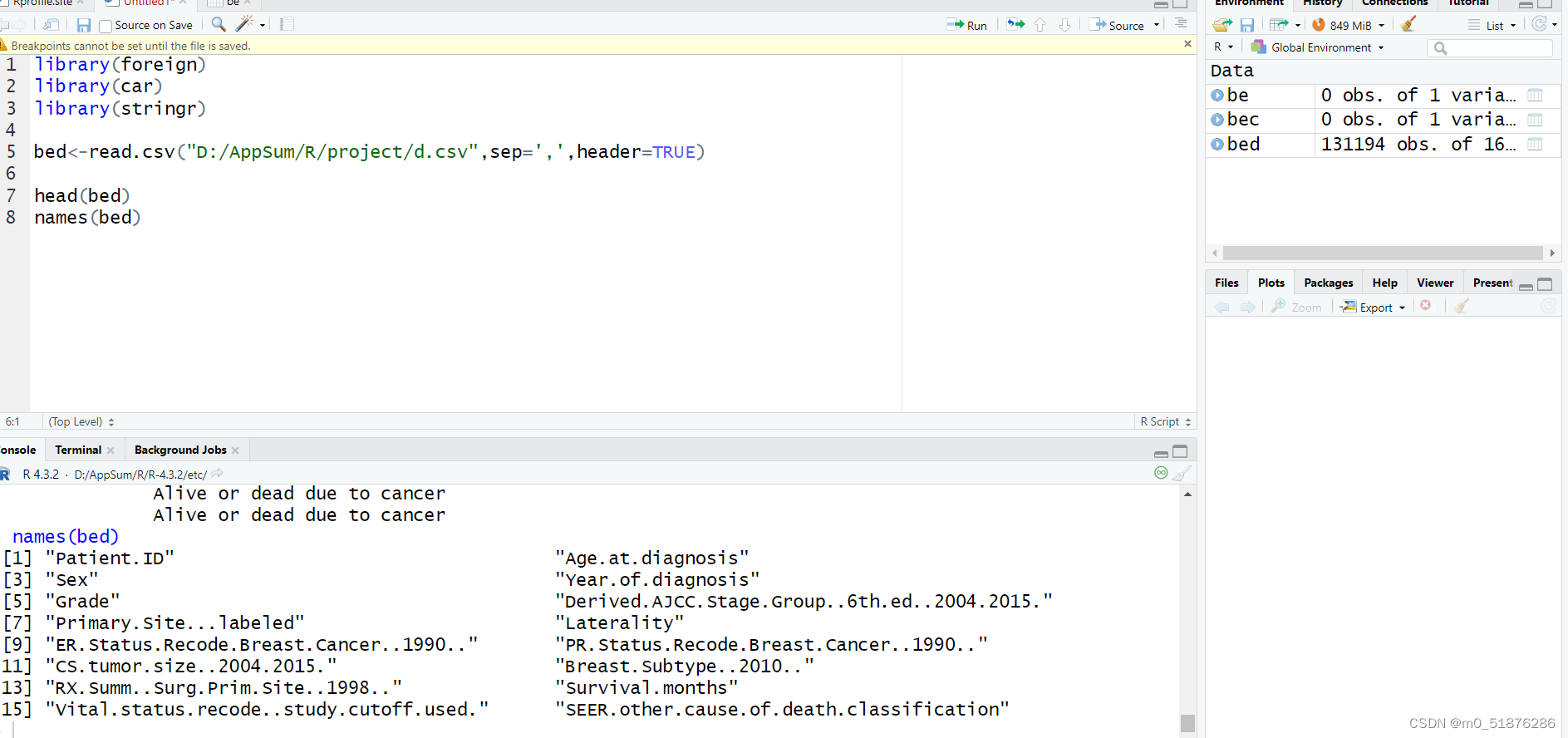
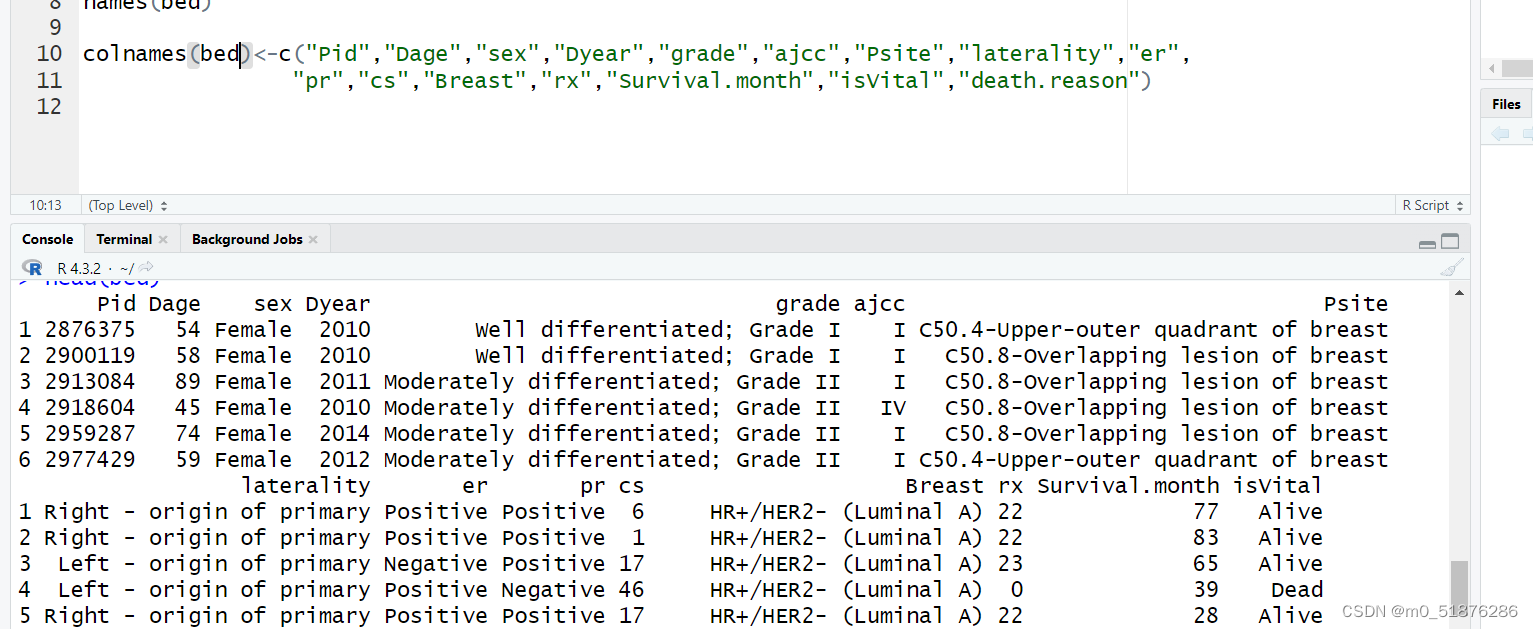
修改标签名称
colnames(bed)<-c("Pid","Dage","sex","Dyear","grade","ajcc","Psite","laterality","er",
"pr","cs","Breast","rx","Survival.month","isVital","death.reason")
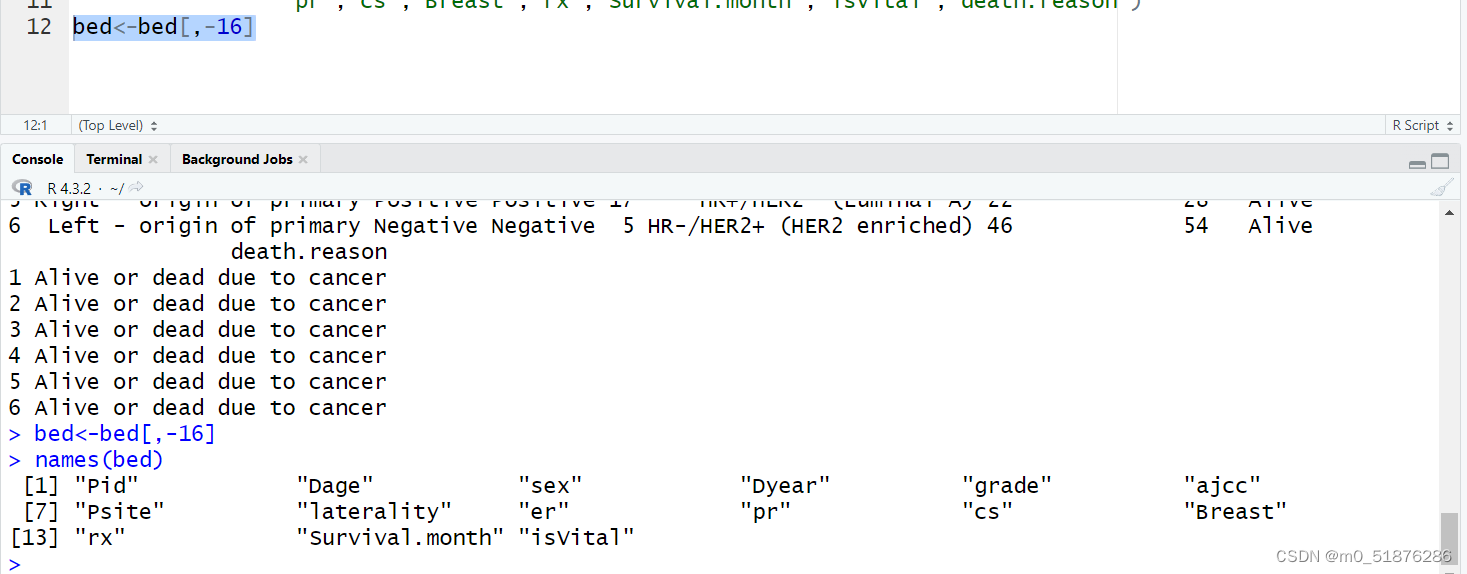
观察数据得到,最后一列标签全是癌症致死,选择删除此列。
bed<-bed[,-16]
通过将字符串转换成数字。
bed$sex<-ifelse(bed$sex=="Female",1,ifelse(bed$sex=="Male",2,NA))
bed$grade<-recode(bed$grade,"'Well differentiated; Grade I'=1;
'Moderately differentiated; Grade II'=2; 'Poorly differentiated; Grade III'=3;
'Undifferentiated; anaplastic; Grade IV'=4;else=NA")#这里是4个分类变量,使用ifelse函数套叠胎麻烦,改用car函数
bed$ajcc<-recode(bed$ajcc,"'I'=1;'II'=2;'III'=3;'IV'=4;else=NA")
bed$Psite<-recode(bed$Psite,"'C50.0-Nipple'=0;'C50.1-Central portion of breast'=1;'C50.2-Upper-inner quadrant of breast'=2;
'C50.3-Lower-inner quadrant of breast'=3;'C50.4-Upper-outer quadrant of breast'=4;
'C50.5-Lower-outer quadrant of breast'=5;'C50.6-Axillary tail of breast'=6;
'7'=7;'C50.8-Overlapping lesion of breast'=8;
'C50.9-Breast, NOS'=9;else=NA")
bed$laterality<-recode(bed$laterality,"'Bilateral, single primary'=1;'Left - origin of primary'=2;
'Only one side - side unspecified'=3;'Paired site, but no information concerning laterality'=4;
'Right - origin of primary'=5;else=NA")
bed$er<-recode(bed$er,"'Borderline'=1;'Negative'=2;'Positive'=3;else=NA")
bed$pr<-recode(bed$pr,"'Borderline'=1;'Negative'=2;'Positive'=3;else=NA")
bed$Breast<-recode(bed$Breast,"'HR-/HER2- (Triple Negative)'=1;
'HR-/HER2+ (HER2 enriched)'=2;'HR+/HER2- (Luminal A)'=3;
'HR+/HER2+ (Luminal B)'=4;else=NA")
bed$isVital<-ifelse(bed$isVital=="Alive",1,ifelse(bed$isVital=="Dead",2,NA))
输出为csv文件即可
write.csv(bed,file = "1.csv")
文章来源:https://blog.csdn.net/m0_51876286/article/details/135524740
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
最新文章
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- 如何用一份完美简历锁定心仪工作?
- 机器人说明书---名词解释031课_python语言_运算符重载
- yarn无法加载文件和‘vue-cli-service‘ 不是内部或外部命令解决方法
- 【python】密码字典生成器
- python接口自动化(十)--post请求四种传送正文方式(详解)
- ECE UN R152 AEBS法规解析
- 揭秘Python的隐秘语法:编程大师的秘密武器
- 计算机网络-物理层基本概念(接口特性 相关概念)
- 一文带你了解:集成电路(IC)从基础概念、制造工艺、种类到广泛应用全解析
- 操作系统第六章输入输出系统课后习题答案