ggkegg玩转KEGG数据 | 富集 | 可视化
本期内容

写在前面
今天分享一个关于KEGG通路图绘制的R包,也许在你后面的分析中可以使用得到。
在KEGG富集分析中,若我们要绘制某一个富集通路,一般回到KEGG官网中寻找该通路的富集图。然后,通过AI,PPT等等一系列手段进行绘制。但是,目前也有一些云平台可以使用,就看自己如何绘制。
今天,分享ggkegg包,一个用于绘制KEGG富集通路的R包,实用性也很强,以及作者提供了详细的帮助文档(PS:我们也基于机器翻译,整理了ggkegg的帮助文档,后面会提供给大家)。
2023教程汇总:2023年教程汇总 | 《小杜的生信笔记》
原文链接:ggkegg | 对KEGG数据进行可视化
加载R包
## 安装R包
BiocManager::install("ggkegg")
# or
devtools::install_github("noriakis/ggkegg")
##--------------------
library(ggkegg)
library(ggfx)
library(igraph)
library(tidygraph)
library(dplyr)
一个简单的例子01
pathway("ko01100") |>
process_line() |>
highlight_module(module("M00021")) |>
highlight_module(module("M00338")) |>
ggraph(x=x, y=y) +
geom_node_point(size=1, aes(color=I(fgcolor),
filter=fgcolor!="none" & type!="line")) +
geom_edge_link0(width=0.1, aes(color=I(fgcolor),
filter=type=="line"& fgcolor!="none")) +
with_outer_glow(
geom_edge_link0(width=1,
aes(color=I(fgcolor),
filter=(M00021 | M00338))),
colour="red", expand=5
) +
with_outer_glow(
geom_node_point(size=1.5,
aes(color=I(fgcolor),
filter=(M00021 | M00338))),
colour="red", expand=5
) +
geom_node_text(size=2,
aes(x=x, y=y,
label=graphics_name,
filter=name=="path:ko00270"),
repel=TRUE, family="sans", bg.colour="white") +
theme_void()

compounds <- c("cpd:C00100", "cpd:C00894", "cpd:C00894", "cpd:C05668",
"cpd:C05668", "cpd:C01013", "cpd:C01013", "cpd:C00222",
"cpd:C00222", "cpd:C00024")
g <- pathway("ko00640") |> mutate(mod=highlight_set_nodes(compounds, how="all"))
ggraph(g, layout="manual", x=x, y=y)+
geom_node_rect(fill="grey",aes(filter=type == "ortholog"))+
overlay_raw_map("ko00640")+
geom_node_point(aes(filter=type == "compound"),
shape=21, fill="blue", color="black", size=2)+
ggfx::with_outer_glow(
geom_node_point(aes(filter=mod, x=x, y=y), color="red",size=2),
colour="yellow",expand=5
)+
theme_void()

g <- pathway("hsa04110")
pseudo_lfc <- sample(seq(0,3,0.1), length(V(g)), replace=TRUE)
names(pseudo_lfc) <- V(g)$name
ggkegg("hsa04110",
convert_org = c("pathway","hsa","ko"),
numeric_attribute = pseudo_lfc)+
geom_edge_parallel2(
aes(color=subtype_name),
arrow = arrow(length = unit(1, 'mm')),
start_cap = square(1, 'cm'),
end_cap = square(1.5, 'cm')) +
geom_node_rect(aes(filter=.data$type == "group"),
fill="transparent", color="red") +
geom_node_rect(aes(fill=numeric_attribute,
filter=.data$type == "gene")) +
geom_node_text(aes(label=converted_name,
filter=.data$type == "gene"),
size=2.5,
color="black") +
with_outer_glow(
geom_node_text(aes(label=converted_name,
filter=converted_name=="PCNA"),
size=2.5, color="red"),
colour="white", expand=4
) +
scale_edge_color_manual(values=viridis::plasma(11)) +
scale_fill_viridis(name="LFC") +
theme_void()

常用例子02
多个微生物基因组评估模块完整性
跨多个微生物基因组评估模块完整性
mod <- module("M00009")
query <- sample(attr(mod, "definition_components"), 5) |>
strsplit(":") |>
sapply("[",2)
query
#> [1] "K01677" "K00164" "K00247" "K00240" "K00246"
mod |>
module_completeness(query) |>
kableExtra::kable()
我们可以评估从多个物种基因组中推断出的 KO 的完整性。在这里,我们将从 PATRIC 服务器获得的 MIDAS 流水线注释文件中可用的 EC 编号映射到 KO,并计算随机获得的物种的完整性。
## Load pre-computed KOs, and recursively perform completeness calculation.
mf <- list.files("../")
mf <- mf[startsWith(mf, "M")]
annos <- list()
candspid <- list.files("../species_dir")
candspid <- sample(candspid, 10)
## Obtain EC to KO mapping file from KEGG REST API
mapper <- data.table::fread("https://rest.kegg.jp/link/ec/ko", header=FALSE)
suppressMessages(
for (i in candspid) {
mcs <- NULL
df <- read.table(paste0("../species_dir/",i), sep="\t", header=1)
fid <- paste0("ec:",df[df$ontology=="ec",]$function_id)
kos <- mapper[mapper$V2 %in% fid,]$V1 |> strsplit(":") |> sapply("[",2) |> unique()
for (mid in mf) {
mc <- module_completeness(module(mid, directory="../"),
query = kos)
mcs <- c(mcs, mc$complete |> mean()) ## Mean of blocks
}
annos[[as.character(i)]] <- mcs
}
)
接下来,我们将使用 ComplexHeatmap 和 simple yEnrich 来可视化结果。我们将通过简化 Enrich 将模块描述的单词云与热图一起绘制,用于确定的集群。
library(ComplexHeatmap)
## Make data.frame
hdf <- data.frame(annos, check.names=FALSE)
row.names(hdf) <- mf
hdf[is.na(hdf)] <- 0
hdf <- hdf[apply(hdf, 1, sum)!=0,]
## Prepare for word cloud annotation
moddesc <- data.table::fread("https://rest.kegg.jp/list/module", header=FALSE)
## Obtain K-means clustering
km = kmeans(hdf, centers = 10)$cluster
gene_list <- split(row.names(hdf), km)
gene_list <- lapply(gene_list, function(x) {
x[!is.na(x)]
})
annotList <- list()
for (i in names(gene_list)) {
maps <- (moddesc |> dplyr::filter(V1 %in% gene_list[[i]]))$V2
annotList[[i]] <- maps
}
col_fun = circlize::colorRamp2(c(0, 0.5, 1),
c(scales::muted("blue"), "white", scales::muted("red")))
ht1 <- Heatmap(hdf, show_column_names = TRUE,
col=col_fun, row_split=km,
heatmap_legend_param = list(
legend_direction = "horizontal",
legend_width = unit(5, "cm")
),
rect_gp = gpar(col = "white", lwd = 2),
name="Module completeness", border=TRUE,
column_names_max_height =unit(10,"cm"))+
rowAnnotation(
keywords = simplifyEnrichment::anno_word_cloud(align_to = km,
term=annotList,
exclude_words=c("pathway","degradation",
"biosynthesis"),
max_words = 40,fontsize_range = c(5,20))
)
ht1

例子03
通过使用 ggforce,可以绘制多个图表来显示哪些基因属于哪个网络。
kne3 <- network("N00485") ## EBV
kne4 <- network("N00030") ## EGF-EGFR-RAS-PI3K
three <- kne3 |> network_graph()
four <- kne4 |> network_graph()
gg <- Reduce(function(x,y) graph_join(x,y, by="name"), list(one, two, three, four))
coln <- gg |> activate(nodes) |> data.frame() |> colnames()
nids <- coln[grepl("network_ID",coln)]
net <- plot_kegg_network(gg)
for (i in nids) {
net <- net + ggforce::geom_mark_hull(alpha=0.2, aes(group=.data[[i]],
fill=.data[[i]], x=x, y=y, filter=!is.na(.data[[i]])))
}
net + scale_fill_manual(values=viridis::plasma(4), name="ID")

例子04
## Numeric vector (name is SYMBOL)
vinflfc <- vinf$log2FoldChange |> setNames(row.names(vinf))
g |>
## Use graphics_name to merge
mutate(grname=strsplit(graphics_name, ",") |> vapply("[", 1, FUN.VALUE="a")) |>
activate(edges) |>
mutate(summed = edge_numeric_sum(vinflfc, name="grname")) |>
filter(!is.na(summed)) |>
activate(nodes) |>
mutate(x=NULL, y=NULL, deg=centrality_degree(mode="all")) |>
filter(deg>0) |>
ggraph(layout="nicely")+
geom_edge_parallel(aes(color=summed, width=summed,
linetype=subtype_name),
arrow=arrow(length=unit(1,"mm")),
start_cap=circle(2,"mm"),
end_cap=circle(2,"mm"))+
geom_node_point(aes(fill=I(bgcolor)))+
geom_node_text(aes(label=grname,
filter=type=="gene"),
repel=TRUE, bg.colour="white")+
scale_edge_width(range=c(0.1,2))+
scale_edge_color_gradient(low="blue", high="red", name="Edge")+
theme_void()

例子05-绘制通路富集全局图
使用 ko01100中的默认值和计算程度来可视化整个全局地图。
ggraph(g2, layout="fr")+
geom_edge_link0(aes(color=I(fgcolor)), width=0.1)+
geom_node_point(aes(fill=I(fgcolor), size=Degree), color="black", shape=21)+
theme_graph()

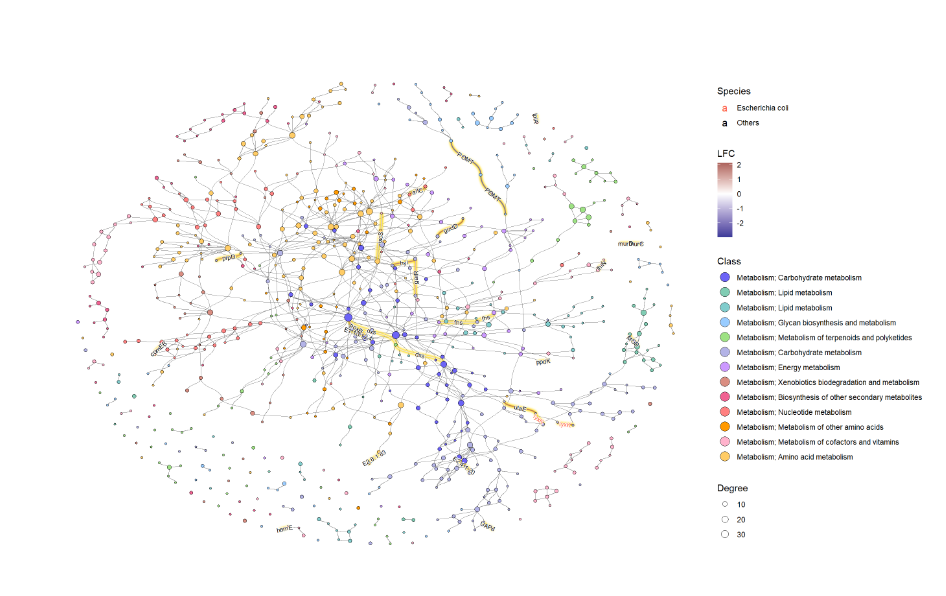
为了有效地进行可视化,我们可以在 KEGG 路径中的各个组件上应用各种宝石图。在这个例子中,我们通过 ggfx 突出显示了由其 LFC 着色的重要边(KO) ,点大小对应于网络中的度,并且我们显示了重要 KO 名称的边标签。KO 名称由“物种”属性着色。这次我们把这个设置为大肠桿菌和其他。
ggraph(g2, layout="fr") +
geom_edge_diagonal(color="grey50", width=0.1)+ ## Base edge
ggfx::with_outer_glow(
geom_edge_diagonal(aes(color=kolfc,filter=siglgl),
angle_calc = "along",
label_size=2.5),
colour="gold", expand=3
)+ ## Highlight significant edges
scale_edge_color_gradient2(midpoint = 0, mid = "white",
low=scales::muted("blue"),
high=scales::muted("red"),
name="LFC")+ ## Set gradient color
geom_node_point(aes(fill=bgcolor,size=Degree),
shape=21,
color="black")+ ## Node size set to degree
scale_size(range=c(1,4))+
geom_edge_label_diagonal(aes(
label=kon,
label_colour=Species,
filter=siglgl
),
angle_calc = "along",
label_size=2.5)+ ## Showing edge label, label color is Species attribute
scale_label_colour_manual(values=c("tomato","black"),
name="Species")+ ## Scale color for edge label
scale_fill_manual(values=hex,labels=class,name="Class")+ ## Show legend based on HEX
theme_graph()+
guides(fill = guide_legend(override.aes = list(size=5))) ## Change legend point size
## Subset and do the same thing
g2 |>
morph(to_subgraph, siglgl) |>
activate(nodes) |>
mutate(tmp=centrality_degree(mode="all")) |>
filter(tmp>0) |>
mutate(subname=compn) |>
unmorph() |>
activate(nodes) |>
filter(bgcolor=="#B3B3E6") |>
mutate(Degree=centrality_degree(mode="all")) |> ## Calculate degree
filter(Degree>0) |>
ggraph(layout="fr") +
geom_edge_diagonal(color="grey50", width=0.1)+ ## Base edge
ggfx::with_outer_glow(
geom_edge_diagonal(aes(color=kolfc,filter=siglgl),
angle_calc = "along",
label_size=2.5),
colour="gold", expand=3
)+
scale_edge_color_gradient2(midpoint = 0, mid = "white",
low=scales::muted("blue"),
high=scales::muted("red"),
name="LFC")+
geom_node_point(aes(fill=bgcolor,size=Degree),
shape=21,
color="black")+
scale_size(range=c(1,4))+
geom_edge_label_diagonal(aes(
label=kon,
label_colour=Species,
filter=siglgl
),
angle_calc = "along",
label_size=2.5)+ ## Showing edge label
scale_label_colour_manual(values=c("tomato","black"),
name="Species")+ ## Scale color for edge label
geom_node_text(aes(label=stringr::str_wrap(subname,10,whitespace_only = FALSE)),
repel=TRUE, bg.colour="white", size=2)+
scale_fill_manual(values=hex,labels=class,name="Class")+
theme_graph()+
guides(fill = guide_legend(override.aes = list(size=5)))

ggkegg更多及更详细的用法,请看:https://noriakis.github.io/software/ggkegg/index.html
为了方便的使用,我们基于机器翻译,将ggkegg帮助文档进行翻译。译文后续将提供给大家。
往期文章:
1. 复现SCI文章系列专栏
2. 《生信知识库订阅须知》,同步更新,易于搜索与管理。
3. 最全WGCNA教程(替换数据即可出全部结果与图形)
4. 精美图形绘制教程
5. 转录组分析教程
一个转录组上游分析流程 | Hisat2-Stringtie
小杜的生信筆記 ,主要发表或收录生物信息学的教程,以及基于R的分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- Linux系统中安装docker-ce
- Vue3中watch和watchEffect的用法
- C#MVC项目---登录
- React快速入门(一)基础与语法
- 《系统架构设计师教程(第2版)》第4章-信息安全技术基础知识-04-信息安全的抗攻击技术
- 大创项目推荐 深度学习疫情社交安全距离检测算法 - python opencv cnn
- Typora Mac激活
- 计算机组成与体系结构(6分)
- 安卓查看进程groups
- CRS-1726 Process failed to run in real-time priority