Cell 文章图复现
发布时间:2024年01月04日
多组差异火山图复现
参考文章: A Spatiotemporal Organ-Wide Gene Expression and Cell Atlas of the Developing Human Heart Figure 2. H
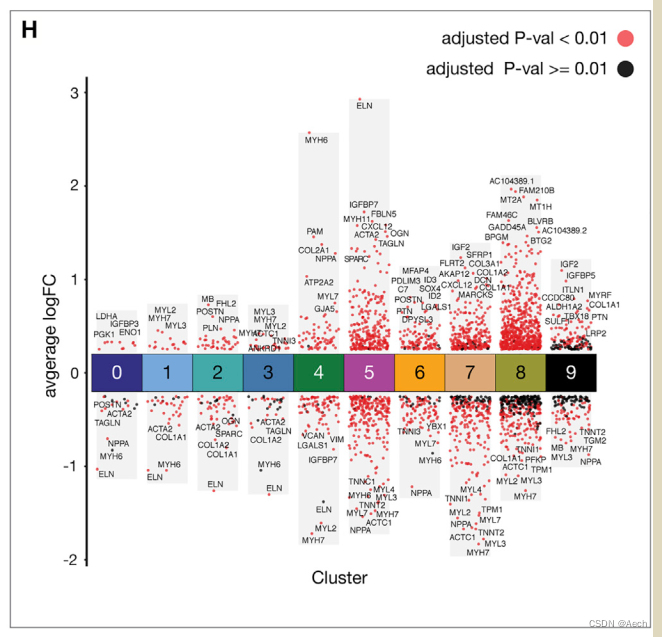
图里主要是单细胞数据不同cluster之间的差异火山图, 所以说白了就是散点图和柱状图的结合, 散点图用差异基因绘制, 柱状图利用logFC最大最小值绘制就完了.
加载包
> library(tidyverse)
> library(ggplot2)
> library(ggpubr)
> library(RColorBrewer)
> library(openxlsx)
> library(ggsci)
> library(ggrepel)
> # Create color parameters
> qual_col_pals = brewer.pal.info[brewer.pal.info$category == 'qual',]
> col_vector = unlist(mapply(brewer.pal, qual_col_pals$maxcolors, rownames(qual_col_pals)))
>
读取数据
> deg <- read.csv("./Differentially_Expressed_Markers_Each_Cluster.csv", header = T)
> deg$cluster <- as.factor(deg$cluster)
> head(deg)
X p_val avg_log2FC pct.1 pct.2 p_val_adj cluster gene
1 1 0 2.558924 0.982 0.289 0 0 DEFB1
2 2 0 2.365316 0.963 0.220 0 0 HMGCS2
3 3 0 2.317304 0.991 0.513 0 0 ATP1B1
4 4 0 2.207154 0.963 0.231 0 0 AC015522.1
5 5 0 2.153153 0.912 0.244 0 0 HSD11B2
6 6 0 2.125726 0.811 0.209 0 0 PAPPA2
> deg <- deg %>% dplyr::filter(p_val_adj < 0.05) %>%
+ dplyr::filter(abs(avg_log2FC) > 0.75) %>%
+ dplyr::select(avg_log2FC, p_val_adj, cluster, gene) # filter and tidy the matrix
>
添加一些注释信息, 例如legend, 上下调, 需要显示名称的基因等
> deg <- deg %>%
+ mutate(label = ifelse(p_val_adj < 0.01, "adjusted P-val < 0.01", "adjusted P-val >= 0.01")) %>%
+ mutate(Change = ifelse(avg_log2FC > 0.75, "UP", "DOWN"))
>
> bardata <- deg %>% dplyr::select(cluster, avg_log2FC ) %>%
+ group_by(cluster) %>%
+ summarise_all(list(tail = min, top = max)) #
> head(bardata)
# A tibble: 6 × 3
cluster tail top
<fct> <dbl> <dbl>
1 0 -5.61 2.56
2 1 -5.13 4.32
3 2 -5.46 2.53
4 3 -4.84 4.81
5 4 -5.60 3.97
6 5 -4.59 2.96
>
> tagedgene <- deg %>% group_by(cluster) %>%
+ slice_max(abs(avg_log2FC), n = 3)
> head(tagedgene)
# A tibble: 6 × 6
# Groups: cluster [2]
avg_log2FC p_val_adj cluster gene label Change
<dbl> <dbl> <fct> <chr> <chr> <chr>
1 -5.61 0 0 ALDOB adjusted P-val < 0.01 DOWN
2 -5.46 0 0 HSPA1A adjusted P-val < 0.01 DOWN
3 -5.09 0 0 GPX3 adjusted P-val < 0.01 DOWN
4 -5.13 0 1 DEFB1 adjusted P-val < 0.01 DOWN
5 -4.61 0 1 CRYAB adjusted P-val < 0.01 DOWN
6 -4.36 1.07e-43 1 ALDOB adjusted P-val < 0.01 DOWN
>
绘制图形
- 利用bardata绘制背景柱状图
ggplot(deg, aes(x = cluster, y = avg_log2FC ))+
geom_col(data = bardata, mapping = aes(x = cluster, y = tail),
fill = "grey", width = 0.8) +
geom_col(data = bardata, mapping = aes(x = cluster, y = top),
fill = "grey", width = 0.8)
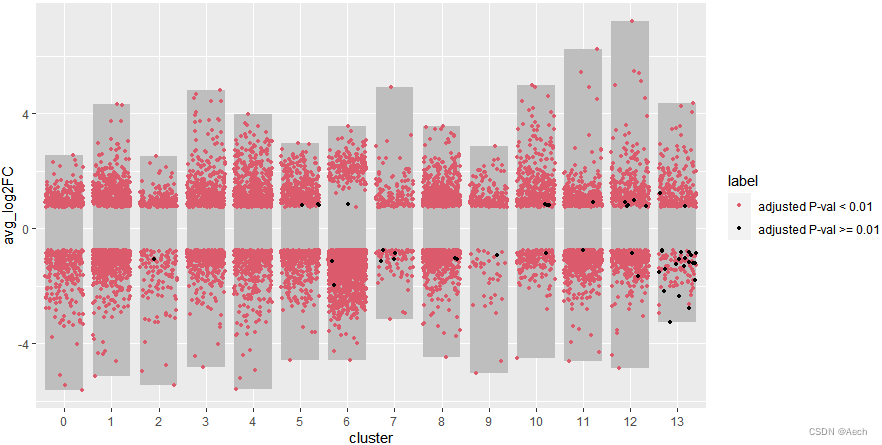
- 添加上散点图, 黑色点有点少了,
不过无所谓能看到就行
ggplot(deg, aes(x = cluster, y = avg_log2FC ))+
geom_col(data = bardata, mapping = aes(x = cluster, y = tail),
fill = "grey", width = 0.8) +
geom_col(data = bardata, mapping = aes(x = cluster, y = top),
fill = "grey", width = 0.8) +
geom_jitter(aes(color = label), size = 1,
position = position_jitter(seed = 0328)) +
scale_color_manual(values = c("#db5a6b", "black"))
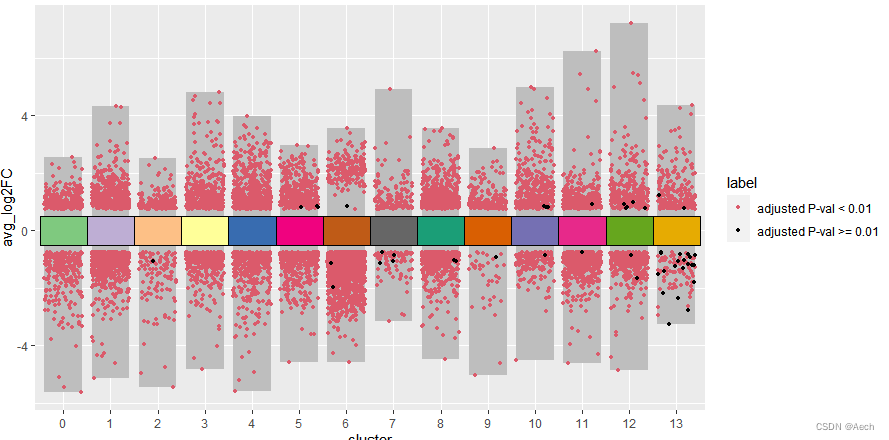
- 添加注释方块
ggplot(deg, aes(x = cluster, y = avg_log2FC ))+
geom_col(data = bardata, mapping = aes(x = cluster, y = tail),
fill = "grey", width = 0.8) +
geom_col(data = bardata, mapping = aes(x = cluster, y = top),
fill = "grey", width = 0.8) +
geom_jitter(aes(color = label), size = 1,
position = position_jitter(seed = 0328)) +
scale_color_manual(values = c("#db5a6b", "black")) +
geom_tile(aes(y = 0, fill = cluster), show.legend = F,
color = "black", width = 1) +
scale_fill_manual(values = col_vector)
- 给想要展示的基因和注释方块添加文字
- 看着有点挤, 点击zoom放大就好了
ggplot(deg, aes(x = cluster, y = avg_log2FC ))+
geom_col(data = bardata, mapping = aes(x = cluster, y = tail),
fill = "grey", width = 0.8) +
geom_col(data = bardata, mapping = aes(x = cluster, y = top),
fill = "grey", width = 0.8) +
geom_jitter(aes(color = label), size = 1,
position = position_jitter(seed = 0328)) +
scale_color_manual(values = c("#db5a6b", "black")) +
geom_tile(aes(y = 0, fill = cluster), show.legend = F,
color = "black", width = 1) +
scale_fill_manual(values = col_vector) +
geom_text(aes(y = 0, label = cluster)) +
geom_text_repel(data = deg %>% filter(gene %in% unique(tagedgene$gene)),
aes(label = gene), position = position_jitter(seed = 0328),
arrow = arrow(angle = 30, length = unit(0.05, "inches"),
ends = "last", type = "open"))

- 最后处理一下背景啥的
ggplot(deg, aes(x = cluster, y = avg_log2FC ))+
geom_col(data = bardata, mapping = aes(x = cluster, y = tail),
fill = "grey", width = 0.8) +
geom_col(data = bardata, mapping = aes(x = cluster, y = top),
fill = "grey", width = 0.8) +
geom_jitter(aes(color = label), size = 1,
position = position_jitter(seed = 0328)) +
scale_color_manual(values = c("#db5a6b", "black")) +
geom_tile(aes(y = 0, fill = cluster), show.legend = F,
color = "black", width = 1) +
scale_fill_manual(values = col_vector) +
geom_text(aes(y = 0, label = cluster)) +
geom_text_repel(data = deg %>% filter(gene %in% unique(tagedgene$gene)),
aes(label = gene), position = position_jitter(seed = 0328),
arrow = arrow(angle = 30, length = unit(0.05, "inches"),
ends = "last", type = "open")) +
theme_minimal() +
theme(axis.line.y = element_line(color = "black", linewidth = 1),
axis.line.x = element_blank(),
axis.text.x = element_blank(),
panel.grid = element_blank(),
legend.title = element_blank())
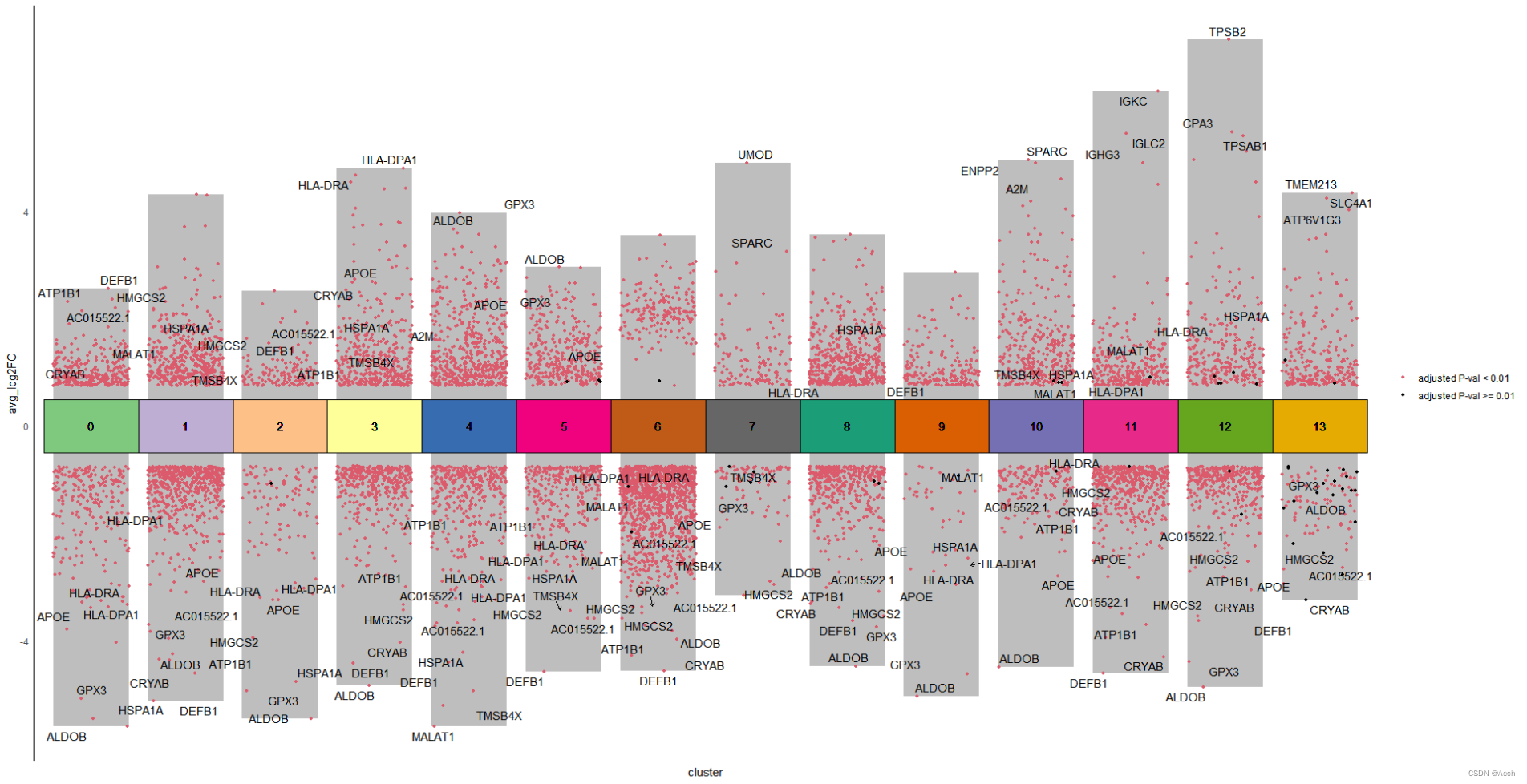
是不是很简单啊 😃
其实不只是单细胞, RNAseq等技术的差异基因也可以组合成类似的矩阵之后绘制相同的多组差异火山图. 理解这个图是柱状图和散点图的结合就可以灵活的绘制类似的图啦 😃
文章来源:https://blog.csdn.net/Aechh/article/details/135384734
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
最新文章
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- 大模型做实体识别任务的原理
- VM-Linux 桥接网络设置
- 什么是事件冒泡?如何组织事件冒泡
- 如何科学地防范冬季流感
- 【QT+QGIS跨平台编译】之二:【zlib+Qt跨平台编译】(一套代码、一套框架,跨平台编译)
- 无风扇工控机的多功能性和空间效率
- 从零开始学习Web自动化:用Python和Selenium实现网站登录功能!
- 结构化流的介绍
- LT7911D是TYPE-C/DP或者EDP转2 PORT MIPI和LVDS加音频
- JMeter+Grafana+Influxdb搭建可视化性能测试监控平台