AnnData:单细胞和空间组学分析的数据基石
AnnData:单细胞和空间组学分析的数据基石
今天我们来系统学习一下单细胞分析的标准数据类型——AnnData!
AnnData就是有注释的数据,全称是Annotated Data。
AnnData是为了矩阵类型数据设计的,也就是长得和表格一样的数据。比如说我们有
n
n
n个样本(观测值),每个样本用一个
d
d
d维的向量表示,这个向量的每一个维度表示着一个特征。也就是说,这是一个
n
×
d
n \times d
n×d的矩阵。
举个例子,在scRNA-seq数据中,每一行表示一个带有barcode的细胞,每一列表示带有gene id的基因。而且数据还不只这一个矩阵,每个细胞和基因可能会有额外的信息(metadata),比如:①细胞的来源;②基因是否甲基化等等。此外还有一些非结构化的metadata,比如画图用的一些参数(调色板等)。
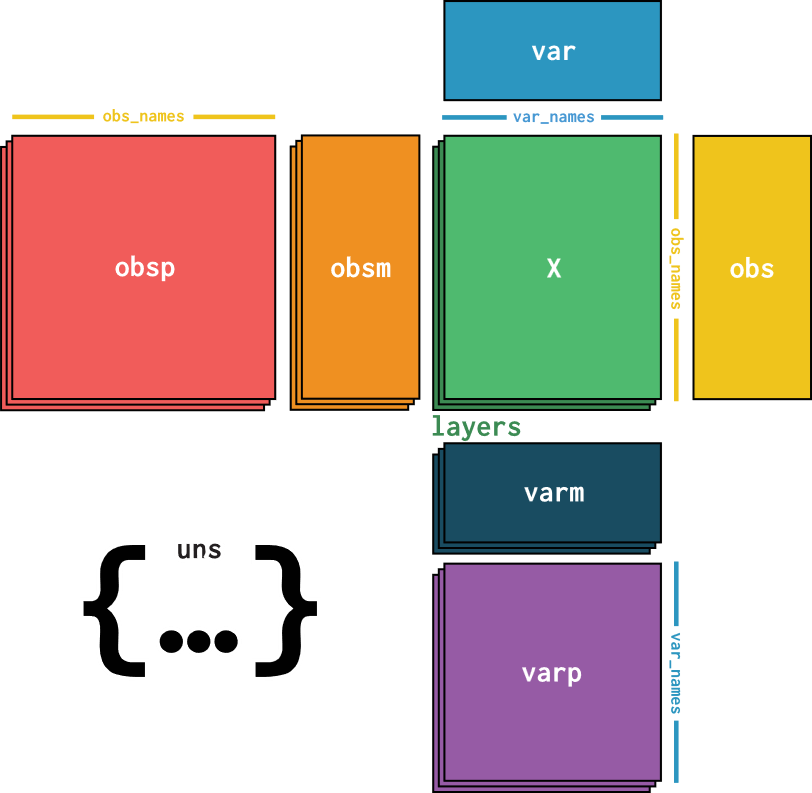
目前来说,似乎除了AnnData,没有其他的数据结构可以做到:
- 处理稀疏性
- 处理非结构化的数据
- 处理样本和特征的metadata
- 容易上手
AnnData的论文也发在了nature biotechnology上!

安装与导入
# !pip install anndata
import numpy as np
import pandas as pd
import anndata as ad
from scipy.sparse import csr_matrix
print(ad.__version__)
0.10.4
初始化AnnData
我们来创建一个AnnData对象吧,首先我们通过NumPy随机生成一些数据,然后再用csr_matrix()转换成稀疏矩阵。
我们生成的数据是一个大小为100*2000数据符合泊松分布的矩阵,可以假装是100个细胞和2000个基因的counts矩阵。
泊松分布(Poisson distribution)是一种离散概率分布,它用于表示在固定时间或空间区间内发生某事件的概率,假设这些事件以已知的恒定平均速率独立发生。泊松分布通常用于模拟那些发生次数较少但有大量独立机会发生的事件。
我们使用泊松分布生成随机数可能是因为我们想模拟一个细胞的基因表达,而这其实也算以某个固定的平均速率发生,而且每个事件的发生是差不多独立的。
泊松分布适合于模拟生物学实验中的计数数据,如RNA测序中每个基因的counts,因为这些counts可以看作是在固定时间内发生的事件数量。
λ=1,代表着事件发生的平均次数是1,这样会比较稀疏
counts = csr_matrix(np.random.poisson(1, size=(100, 2000)), dtype=np.float32)
# 使用NumPy库生成一个形状为100x2000的数组,数组中的每个元素都是从参数为1的泊松分布中随机抽取的数值。
# 将上述生成的数组转换为压缩稀疏行(CSR)格式的稀疏矩阵。
# CSR格式是一种高效存储和操作稀疏矩阵的方式,特别是当矩阵中大部分元素为零时。
adata = ad.AnnData(counts)
# 创建了一个AnnData对象,它是anndata库中的一个核心数据结构,用于存储带注释的多维数据。
# 在这里,counts稀疏矩阵被用作AnnData对象的主数据矩阵X。
adata
AnnData object with n_obs × n_vars = 100 × 2000
上面的输出是总结了一下样本和特征有多少,如果想看我们的数据就可以通过adata.X。
adata.X
<100x2000 sparse matrix of type '<class 'numpy.float32'>'
with 125993 stored elements in Compressed Sparse Row format>
现在我们来设置一下样本和特征的名字,这也算是建立索引的步骤。
# 设置观测值名称,每个名称格式为"Cell_序号"
adata.obs_names = [f"Cell_{i:d}" for i in range(adata.n_obs)]
# 使用列表推导式创建了一个新的列表,列表中的每个元素都是以"Cell_"开头,后面跟着一个整数序号的字符串。
# 这个序号从0开始,一直到adata.n_obs(观测值的数量)减1。
adata.var_names = [f"Gene_{i:d}" for i in range(adata.n_vars)]
print(adata.obs_names[:10])
Index(['Cell_0', 'Cell_1', 'Cell_2', 'Cell_3', 'Cell_4', 'Cell_5', 'Cell_6',
'Cell_7', 'Cell_8', 'Cell_9'],
dtype='object')
取子集
这么多数据,我们一般会关注自己想要的,索引方式非常方便。直接用列表把样本和特征的名字一放就行。
adata[["Cell_1", "Cell_10"], ["Gene_5", "Gene_1900"]]
View of AnnData object with n_obs × n_vars = 2 × 2
加点额外的metadata
样本/特征
细胞有类别、基因也不仅仅只有counts,怎么加额外的特征呢?这也很简单,就搞一个和样本或者特征一样长的向量赋值即可。
So we have the core of our object and now we’d like to add metadata at both the observation and variable levels. This is pretty simple with AnnData, both adata.obs and adata.var are Pandas DataFrames.
ct = np.random.choice(["B", "T", "Monocyte"], size=(adata.n_obs,))
adata.obs["cell_type"] = pd.Categorical(ct) # Categoricals are preferred for efficiency
adata.obs
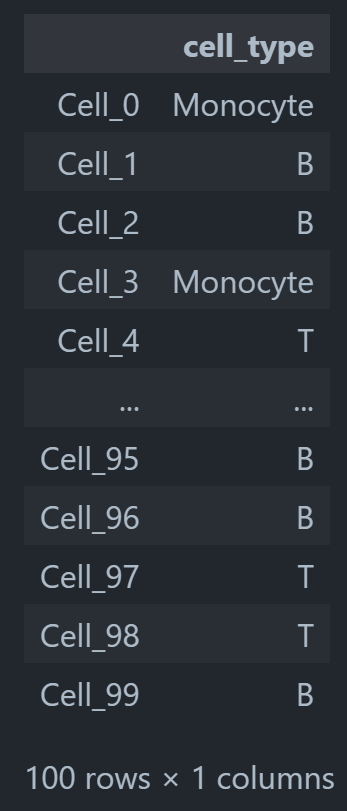
我们可以发现,我们的数据描述也变了,多了一个obs: 'cell_type',这也就是我们刚刚定义的细胞类型。
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
通过metadata取子集
我们可以用我们刚刚为细胞加的特征cell_type来选择我们想要的数据,比如我们想要所有B细胞的数据。
bdata = adata[adata.obs.cell_type == "B"]
bdata
View of AnnData object with n_obs × n_vars = 36 × 2000
obs: 'cell_type'
样本和特征水平的多维度metadata(矩阵)
我们有时候想加的metadata不是一个向量能描述的,它可能是个矩阵。比如细胞的UMAP降维结果,降到2维就是一个细胞个数*2维的矩阵。这时候我们就可以用obsm或者varm来添加就行。
adata.obsm["X_umap"] = np.random.normal(0, 1, size=(adata.n_obs, 2))
# 生成一个100*2的正态分布随机数矩阵
adata.varm["gene_stuff"] = np.random.normal(0, 1, size=(adata.n_vars, 5))
adata.obsm
AxisArrays with keys: X_umap
再看看我们的adata,又更新了。
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
obsm: 'X_umap'
varm: 'gene_stuff'
关于.obsm/.varm的额外说明:
- .obsm和.varm可以存储来自不同来源的矩阵。这些数据可以是Pandas DataFrame、scipy稀疏矩阵或numpy数组。也就是说,我们可以将这些不同类型的数据结构直接存储在.obsm或.varm中,而无需进行转换。
- 当使用scanpy进行单细胞分析时,.obsm和.varm中的值(列)不容易直接用于绘图。相比之下,.obs中的项可以更容易地被绘制在UMAP等降维图上。例如,如果我们有UMAP坐标存储在.obsm中,我们可能需要将它们转移到.obs中,以便使用scanpy的绘图功能。
非结构化的metadata
非结构化,就是随便什么数据都行,不管是列表、字典还是什么其他的,只要是对于我们数据分析有用的都可以放进来,就放在.uns里面就行。
adata.uns["random"] = [1, 2, 3]
adata.uns
OrderedDict([('random', [1, 2, 3])])
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
obsm: 'X_umap'
varm: 'gene_stuff'
欸,非结构化metadata的就没有直接显示。
Layers
最后得说一下layers,这个非常有用。我们经常会对原始数据进行处理,比如标准化、对数化等等。这些可以转换后的数据可以存储在layer中。
adata.layers["log_transformed"] = np.log1p(adata.X)
adata
AnnData object with n_obs × n_vars = 100 × 2000
obs: 'cell_type'
uns: 'random'
obsm: 'X_umap'
varm: 'gene_stuff'
layers: 'log_transformed'
还原回DataFrames
直接使用.to_df()即可,还可以指定layer。
adata.to_df(layer="log_transformed")

你看,dataframe的行列索引也都是我们的.obs_names/.var_names。
保存结果
AnnData提供了一种基于HDF5的持久化文件格式:h5ad。这种格式专门设计用于存储AnnData对象,使得数据可以高效地被读取和写入。
当AnnData对象中有包含少量类别的字符串列时,如果这些字符串列还没有被转换为分类数据类型(categoricals),AnnData会自动将它们转换为分类数据类型。分类数据类型是pandas库中的一种数据类型,用于表示具有固定数量类别的变量,这种类型在内存使用和性能上比普通字符串类型更优。
例如,如果我们有一列,全是字符串,它只包含几个不同的值(如"yes"和"no"),AnnData会自动将这个列的数据类型转换为分类数据类型,这样可以提高数据处理的效率和速度。
adata.write('my_results.h5ad', compression="gzip")
!ls -lh | grep my_results.h5ad
-rw-rw-r-- 1 659K Jan 14 15:31 my_results.h5ad
结语
这次只是学习了AnnData最基础的知识,之后会介绍AnnData进阶的操作。
参考资料:Getting started with anndata — anndata 0.11.0.dev46+g0219fef documentation
如果觉得还不错,记得点赞+收藏哟!谢谢大家的阅读!( ̄︶ ̄)↗
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- HTML5期末大作业:HTML+CSS茶叶官网网页设计实例 企业网站制作
- 十款强大的企业电脑加密软件推荐(企业电脑加密软件哪个最好用)
- FreeRTOS学习--49讲 事件标志位
- 亚马逊站内广告位置在哪设置?怎么设置广告位置?-站斧浏览器
- 如何在Windows 11的桌面中添加此电脑图标,这里提供四种方法
- 洛谷 P3397 地毯 刷题笔记 二维差分矩阵
- 苹果M芯片电脑使用IE浏览器教程
- 05. Springboot admin集成Actuator(一)
- 【每日一题】收集足够苹果的最小花园周长
- epoll并发编程