2024仿制药注册审批流程<一文讲通透>
随着医药科技的进步和创新,仿制药在世界范围内得到了广泛应用。仿制药是指在原研药品专利保护期满后,依据原研药品的临床试验数据和技术文献,通过科学合理的方法,生产出与原研药品具有相同质量、安全性和有效性的药品。仿制药的注册申报是仿制药上市前的重要环节,本文将介绍仿制药注册申报的流程和要求。
简化流程:前期调研→研发阶段→注册资料整理→申报递交和核查审评
- 前期项目调研准备工作:
在进行仿制药注册申报前,申请人需要进行一系列的准备工作。如原研药研发生产厂家、原料药厂商、辅料厂商、药包材、处方组成、专利信息、市场前景等信息调研,调研的内容要全面、精细、时间长,形成调研报告,同时形成项目可行性报告。
调研信息主要包括制剂、分析(质量研究范畴)、市场等内容,详见下表

可通过专业医药数据库工具进行调研会更加高效,如药融云数据库、参比购网、英国EMC的SPC等。
- 仿制药研发阶段工作:
仿制药研发阶段工作可分为:处方研究、小试处方工艺筛选、放大及中试研究、可行批生产研究、工艺验证、注册资料整理、质量标准复核、CDE沟通、注册核查等工作
处方研究内容包括处方筛选、质量信息,见下图

●小试处方工艺筛选(小试研究)
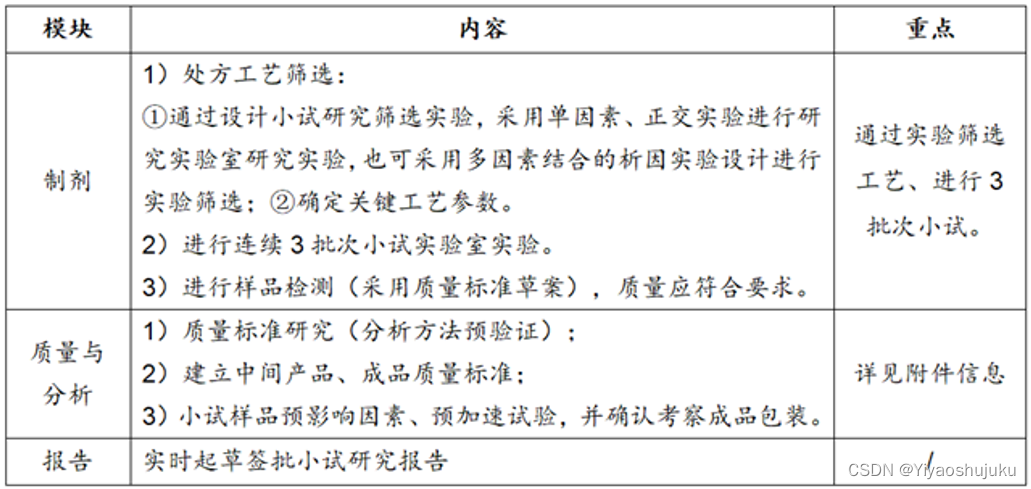
●放大及中试研究

●工艺验证

三、注册资料整理:
申请人需要准备仿制药注册资料的整理。注册资料包括但不限于以下内容:
●注册资料整理
主要资料整理有:①稳定性考察数据汇总整理与总结;②工艺控制、质量控制资料;③产品质量标准研究与质量研究资料等。
主要内容包括:药学研究资料综述、证明性文件资料、生产工艺研究资料及文献、处方及工艺研究资料与文献、质量研究资料、生产信息、质量控制(辅料与成品质量控制)、药效学(药理毒理)研究资料、对照品与杂质谱研究资料、稳定性研究资料等。
四、申报递交和核查审评
●质量标准复核
- 提交药品注册检验申请表/注册申报受理通知书等。
如:化学药品3类、4类上市许可注册申报流程
登录'国家药品监督管理局-政务服务门户'-->右上角“登录”-点击“法人登录”进入登录界面-->点击“账号设置”-“账号绑定”-进入“药品业务应用系统”-->“普通用户登录”-->选择“境内生产药品注册”-->进入后选择“境内生产药品注册上市许可”-->填写申请表-->全部填写完成后,点击“暂存”保存-->暂存后会出现自查表和专利声明,分别填写-->全部信息填写完成、上传附件后,点“暂存”保存,确认不再修改点“申报”正式提交。
2.样品及物品准备:工艺验证3批样品(3倍检验量,至少还有6个月有效期)、对照品(杂质/主成分对照品,3倍检验量)、相关色谱柱等。
3.文件资料:①相关原料药、原辅包材证明性文件资料;②质量标准(最新版)及质量标准对比表;③相关检验方法分析方法验证报告;④3批工艺验证样品企业自检COA。
4.全部纸质资料文件均需盖企业章(准备电子版1份)。
●注册现场核查
1.注册核查:①注册研发现场-药学研制/药理毒理学研制/药物临床试验;②生产现场。
2.目的:核实注册申报资料的真实性、一致性以及药品上市商业化生产条件。
3.重点:申报资料、工艺验证、质量控制、生产现场、辅助文件、人员培训等。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- Focal Loss
- Vue3封装可拖拽的弹窗
- STM32 DAC+串口
- java公交系统毕业论文
- 基于ChatGPT的安卓端语音助手
- 秒杀相关问题及答案(2024)
- 自学笔记Linux--实用技巧--压缩和解压
- String 类的常用方法都有那些?
- FPGA高端项目:Xilinx Artix7 系列FPGA纯verilog图像缩放工程解决方案 提供4套工程源码和技术支持
- 【LabVIEW FPGA入门】模拟输入和模拟输出