原料药开发研发&审批流程(注册必读)
仿制药注册申报简化流程:立项调研-->工艺路线的筛选与确定-->反应参数的优化-->中间体以及粗品的纯化后处理-->API 的结构确证-->杂质研究-->晶型研究-->工艺放大-->公斤级试验以及中试-->资料撰写-->注册申报
(文末附注册申报全流程图&相关费用)
一、立项调研
原料药产品立项调研是为了评估和确定是否值得开展某个原料药产品项目。以下是在原料药产品立项调研中的主要内容:
| 调研内容 | 描述 |
| 市场需求分析 | 了解目标市场的需求情况,包括市场规模、增长趋势、竞争情况等。这可以为后续的技术可行性评估和市场营销策略提供基础。 |
| 技术可行性评估 | 在了解市场需求后,评估所需技术的可行性和成熟度。这有助于确定项目的技术难度和风险,并决定是否继续进行后续调研。 |
| 法规和政策分析 | 了解相关的法规和政策要求,包括药品注册、生产许可、质量控制等方面的要求。这可以帮助评估项目的合规性和可行性,以及确定项目的法规风险。 |
| 成本效益分析 | 在技术和法规方面的评估之后,进行成本效益分析。这有助于评估项目的经济可行性,并决定是否继续推进项目。 |
| 风险评估 | 在对市场需求、技术可行性和法规要求进行评估后,对项目可能面临的风险进行评估。这有助于制定风险管理策略,降低项目失败的概率。 |
| 合作伙伴评估 | 在评估项目的技术、市场和风险后,评估可能的合作伙伴。这有助于确定合作伙伴的可靠性和能力,以支持项目的顺利进行。 |
| 市场营销策略 | 在综合评估市场需求、技术可行性和合作伙伴后,制定适合目标市场的市场营销策略。这有助于确定如何将产品推向市场并获取市场份额。 |
| 时间计划和资源规划 | 在前面的调研基础上,制定项目的时间计划和资源需求。这有助于确保项目能够按计划进行,并合理分配资源。 |
在调研目标市场需求分析时,可通过垂直分析工具进行分析,如药融云-原料药用量推算数据库、原料药情报局(合成路线、海关进出口、供应商、登记注册、用量推算、上市制剂、专利、趋势、质量标准、价格、生产制造商、报告)等,所需时间15到30天。
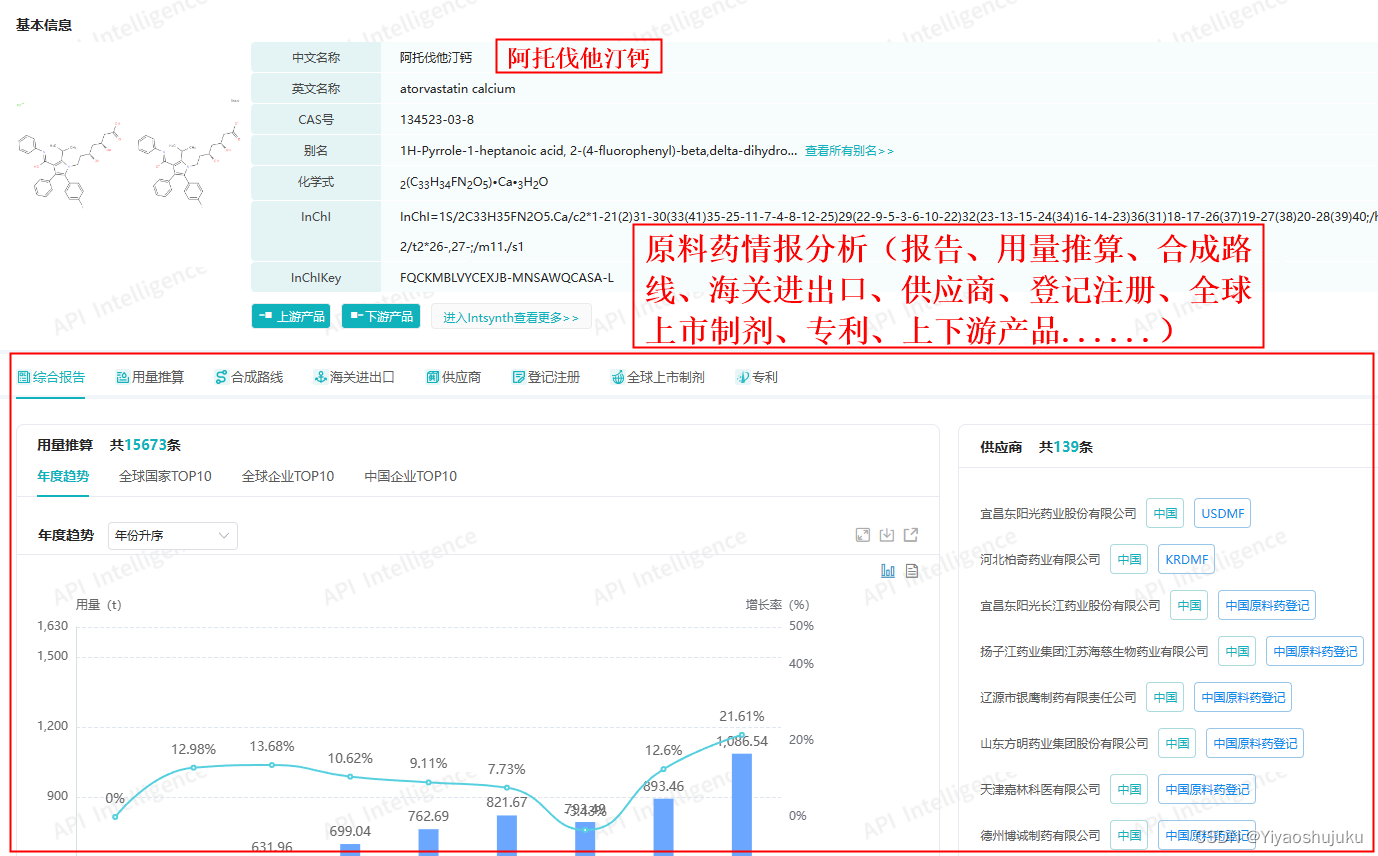 ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? 图源:药融云-原料药情报局(阿托伐他汀钙)
? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? 图源:药融云-原料药情报局(阿托伐他汀钙)
二、工艺路线的筛选与确定
1、合成路线筛选
对每条合成路线进行实验和考察,描述在合成过程中遇到的困难,并分析不同方法的反应机理以及可能产生的杂质。
2、确定产品结构
使用至少1H-NMR和质谱分析来证明合成出的产品与API结构相同,并确保纯度不低于95%。
3、探索中控和API分析方法(如HPLC、TLC)
提供1克以上的API,用于探索各步反应液的分析方法。
4、确定工艺路线
通过对合成路线的筛选,最终确定或暂定准备开发的工艺路线,以进行下一步的工艺优化工作。
预计完成时间:1-1.5个月。
三、反应参数的优化
1、反应机理:主要反应机理和次要反应机理
2、影响因素:溶剂类型、溶剂用量、温度、pH值、物料配比、加料方式、反应时间、无水无氧条件等。
3、选择影响因素的依据:控制杂质含量和产物收率的要求。
突出关键影响因素和关键控制点,并说明每个工艺参数对反应的影响。
- 中间体以及粗品的纯化后处理
优化每个反应步骤的后处理条件,仅采用工业上可行的方法,如洗涤、萃取和结晶。列出可行的后处理方法,并进行比较,选择操作简便、收率高、纯度好的后处理方法。
中间体的纯化:
首先判断是否需要对中间体进行纯化,并说明理由,然后选择适当的方法。
粗品的纯化:
①洗涤:确定洗涤次数、判断洗涤终点,确定洗涤溶剂的用量等。
②萃取:选择合适的萃取溶剂和用量,确定萃取次数等。
③结晶(注意晶型):选择适当的结晶溶剂、结晶方式和优化结晶条件。
在初步确定每个反应步骤和后处理条件后,同时继续优化后续反应和后处理过程,并逐步放大规模,至少达到百克级。
在所有小试工艺和后处理方法初步确定后,进行三批百克级的小试实验,以验证小试条件的稳定性,为后续的放大和公斤级试验做准备。
提供至少50克以上纯度超过99%、单杂质小于0.5%的API,以及每个中间体至少20克以上纯度大于95%。
(三+四)预计完成时间:3-5个月。
五、API 的结构确证
根据API的分子结构和相关文献,可以采用以下方法和方案进行结构确证,并解释这些方法的测试目的。至少包括以下测试内容:高分辨质谱(或元素分析)、红外光谱、核磁共振氢谱、核磁共振碳谱、DEPT碳谱、H-H COSY、C-H COSY,其余的热分析、XRD、旋光度、圆二色谱等根据具体情况而定。
| 测试项目 | 对照品需求 | 测试目的(具体) | |
| 1 | 高分辨质谱 | 否 | 结构难以进行元素分析,保证纯度下可获得元素组成的相关信息。 |
| 2 | 元素分析 | 否 | 可获得药物的元素种类及含量,与理论值误差不得超过3%。 |
| 3 | 红外光谱 | 是 | 可推测出可能存在的化学键、官能团及初步连接方式,亦可给出药物的几何构型、晶型、立体构象等。 |
| 4 | 紫外光谱 | 是 | 可获得结构中可能含有的发色团、助色团种类以及初步的连接方式。 |
| 5 | 1H NMR | 否 | 可获得结构中氢原子数目、周围化学环境、相互间关系、空间排列等信息 |
| 6 | H-H COSY | 否 | 氢原子之间相互耦合的信息 |
| 7 | 13C NMR | 否 | 可获得不同碳原子的类型以及所处的不同化学环境信息。 |
| 8 | DEPT碳谱 | 否 | 进一步明确区分碳原子的类型 |
| 9 | C-H COSY, HMQC | 否 | 氢原子与相连碳的信息 |
| 10 | C-H COSY, HMBC | 否 | 氢原子与3个键之内的碳的信息 |
| 11 | ORD旋光光谱 | 是 | 通过比较药物的旋光性,可得到手性药物的相对构型信息 |
| 12 | CD圆二色性光谱 | 是 | 测定物质在圆偏正光下的Cotton效应,获得结构中发色团周围环境的立体化学信息,与一个绝对构型已知的相似化合物比较,推导出待测物的绝对构型 |
| 13 | DSC差热分析 | 是 | 可获得药物的吸附水/溶剂、结晶水/溶剂以及熔点、有无多晶型存在和热焓值等信息 |
| 14 | TGA热重分析 | 是 | 可获得药物的吸附水/溶剂、结晶水/溶剂及初步的分解温度等信息 |
| 15 | XRSD粉末衍射 | 是 | 用于固态单一化合物的鉴别与晶型确定,晶态与非晶态物质的判断,原料药晶型的稳定性研究等 |
| 16 | XRPD单晶衍射 | 否 | 可获得药物晶型的相关信息、药物的相对或绝对构型以及与药物以结晶形式存在的水/溶剂及含量等一系列信息 |
| 17 | MS | 否 | 用于原子量和分子量的测定、同位素分析、定性或定量的分析? |
手性药物的结构确证可以采用以下方法:X射线单晶衍射(XRSD)、核磁共振(NMR)、圆二色谱(CD)、旋光色散(ORD)以及核Overhauser效应谱(NOESY或NOE谱)。
晶型测定方法包括:X射线粉末衍射(XRPD)、红外光谱(IR)、熔点测定、热分析和光学显微镜等。
对于结晶水或结晶溶剂的分析,可以使用热重分析、差热分析、干燥失重、水分测定和X射线单晶衍射等方法。
金属原子以及F、P等元素的测定可以通过原子发射光谱(AES)和原子吸收光谱(AAS)进行。AES用于金属元素的定性研究,而AAS则用于金属元素的定量研究。
综合解析是将各种谱图和数据之间的有机联系进行综合分析,以相互佐证化合物的结构。
六、杂质研究
杂质列举
1.1 溶剂残留:列举一些不常见、高毒性、基因毒性的溶剂,例如二氯甲烷、四氯化碳、甲醇、乙醇等,并说明它们的来源,如合成过程中的溶剂、反应物的纯度等。
1.2 无机杂质:列举特别的、高毒性的无机杂质,如砷盐、硫化物、亚硫酸氢盐、氰化物等,并解释它们产生的原理,例如反应条件、原料纯度等。常见的氯化物、硫酸盐等无机杂质不再列举。
1.3 有机杂质:列举反应过程、中间体和成品中的所有有机杂质,包括副反应产物、未反应的起始物质、杂质引入的可能来源等。同时,提供这些杂质的鉴定方法,并解释它们产生的机理。
杂质的影响因素以及控制方法
2.1 溶剂残留:针对不常见、高毒性、基因毒性的溶剂,提供数据证明溶剂残留的控制方法,如洗涤次数、洗涤时间、干燥温度、干燥时间、真空度等。
2.2 无机杂质:针对不常见、高毒性的无机杂质,提供数据证明它们的控制方法。
2.3 有机杂质:
2.3.1 各步反应过程、中间体中的杂质控制:详细说明最后一步反应之前所有合成步骤和中间体中杂质的影响因素,并提供控制方法。最好使用图表等直观方式展示,同时明确说明后处理前后的杂质变化情况。
2.3.2 粗产品中的杂质控制:详细说明最后一步反应中杂质的影响因素,并提供控制方法。同样,使用图表等方式展示后处理前后的杂质变化情况。
2.3.3 API精制品中有关物质的控制:详细描述精制工艺开发过程,以及精制前后有关物质的变化情况,并提供完整的数据证明精制过程的可靠性。
有关物质的合成、分离、纯化
提供有关物质的获取方法,包括合成方法、柱层析、制备薄层色谱(TLC)、制备高效液相色谱(HPLC)等。提供不少于0.1g的样品,并确保纯度不低于99%,其他单个杂质的含量不得超过0.5%。
杂质的结构确证(参见API的结构确证)
根据API的结构确证方法,进行杂质的结构确证工作。
预计完成时间为1-1.5个月。
七、晶型研究
1、晶型相关文献综述
提供完整、详细的文献综述,包括与晶型相关的研究论文和专利内容。介绍不同晶型的种类、鉴别方法以及制备方法。
2、晶型选择的依据
详细介绍被仿产品的晶型及相关测试数据,并解释自主开发产品晶型选择的依据和理由。
3、晶型的控制
介绍晶型控制的关键因素,包括结晶条件的选择,如结晶溶剂种类、溶剂用量、结晶温度和结晶时间等。
4、晶型测定方法
提供多种晶型测定方法,包括外观观察、熔点测定、红外光谱、热重分析(TGA)、差示扫描量热法(DSC)、X射线粉末衍射(XRD)、X射线单晶衍射、光学显微镜等。
预计完成时间为1-1.5个月。
八、工艺放大
基于前述的实验结果,初步确定了工艺的关键参数,包括温度、时间、物料选择、终点判断以及反应现象等。通过将实验规模从100克逐步放大至500克、公斤级,不断调整工艺参数,以获得三批稳定且符合质量标准的产品。在这个过程中,要注意对杂质的变化进行监测和控制,并记录产率的变化情况。一旦获得稳定的中试产品,接下来需要制定详细的小试工艺放大操作方法或编写实验报告。
九、公斤级试验以及中试
在进行公斤级试验时,根据杂质和产率的变化,进行参数调整,以确定是否需要补充一些小试验的数据。通过不断调整工艺参数,直至获得三批稳定且符合质量标准的产品。
一旦获得稳定的公斤级产品,需要制定更为详细的工艺放大实验报告,预计完成时间为1-2个月。
十、资料撰写
将以上所有数据进行收集整理,对申报资料原料药药学部分进行撰写,依照CTD要求。
十一、注册申报
根据《药品注册管理办法》27号令,药品审评中心(CDE)在审评制剂申报时,可对制剂所用的化学原料药进行关联审评审批。此外,对于仿制境内已上市制剂所用的化学原料药的,申请人可申请单独审评审批。其审评流程如下所示:
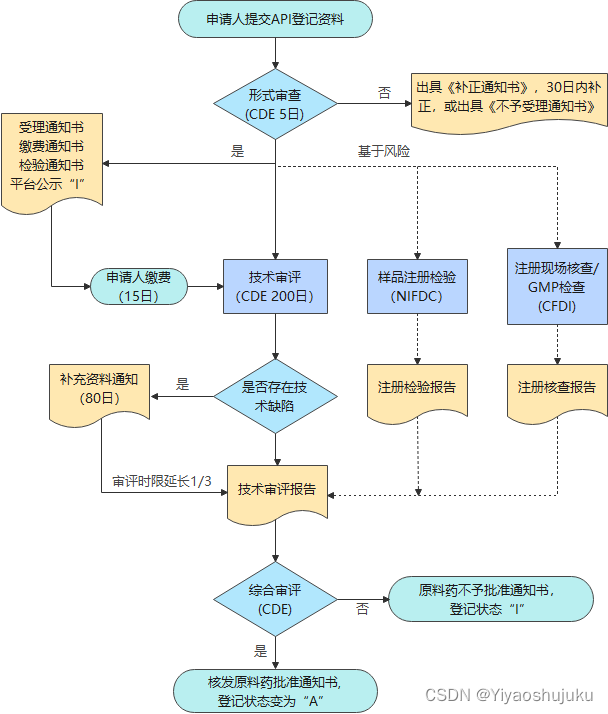
? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? 仿制境内已上市化学原料药审批流程图
附:相关收费标准
根据《药品注册收费标准》知晓各类原料药缴费金额如下:
| 原料药类型 | 缴费金额 |
| 新药申报所关联的境内生产化学原料药 | 21.60万 |
| 新药申报所关联的境外生产化学原料药 | 29.695万 |
| 仿制药申报所关联的/单独申报的境内生产化学原料药 | 18.36万 |
| 仿制药申报所关联的/单独申报的境外生产化学原料药 | 36.76万 |
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- uni-app 中使用定时器和取消定时器
- 车载数字钥匙应用芯片方案
- 第10章-第2节-Java多线程中的synchronized锁
- 全相联映射,组相连映射直接相连映射题
- uni-app小程序的微信一键登录,如何把判断是否签署协议放在手机号授权之前。
- Linux:centos yum安装指令指南
- QT中的信号与槽的讲解
- anaconda prompt进入虚拟环境 打开spyder
- Qt拖拽事件简单实现
- apisix 插件配置 未生效 未起作用