【Gene Expression Prediction】Part1 基因表达数据的获取与分析
文章目录
来自Manolis Kellis教授(MIT计算生物学主任)的课
YouTube:(Gene Expression Prediction - Lecture 09 - Deep Learning in Life Sciences (Spring 2021)
Slides: slides
本节课分为四个部分,本篇笔记是第一部分。
本节主要是介绍如何获得和分析基因表达数据。主要是为了后面的三个讲座铺垫。首先,探讨不同的方法和技术来获取基因表达数据。之后学习如何分析这些数据,包括上采样方法来解决数据不平衡问题,以及压缩感知技术来处理高维数据。最后,我们会讨论如何预测RNA剪接。
Gene Expression Prediction

- 介绍(Intro):
- 这部分可能会介绍基因表达的基础知识、无监督学习的概念以及聚类技术。
- 上采样(Up-sampling):
- 讨论如何从已知的1000个基因的表达数据预测大约20,000个基因的表达水平,这可能涉及到机器学习技术和统计推断。
- 压缩感知(Compressive sensing):
- 探讨如何使用压缩感知技术从综合测量中预测基因表达,这是一种能够从少量观测数据中重建未知信号的技术。
- DeepChrome+LSTMs:
- 介绍一种结合了深度学习(DeepChrome模型)和长短期记忆网络(LSTMs)的方法来预测染色质数据中的基因表达。
- 预测剪接从序列(Predicting splicing from sequence):
- 说明如何使用成千上万的特征从DNA序列预测剪接事件,剪接是基因表达调控的一个关键过程。
- 客座讲座:Flynn Chen, Mark Gerstein实验室, 耶鲁大学:
- Flynn Chen将讨论如何从染色质特征预测报告基因的表达。
- 客座讲座:Xiaohui Xie, 加州大学欧文分校:
- Xiaohui Xie将讲述如何从部分子集抽样预测基因表达,以及如何进行多组学整合的表示学习。
- 客座讲座:Kyle Kai-How Farh, Illumina:
- Kyle Kai-How Farh将探讨如何从序列预测剪接。
1. Intro
在另一篇博客中:基因表达分析聚类&分析
2. Up-sampling
基因表达测量方面的"up-sampling"。其中两个应用:数字信号放大(Digital signal upscaling)和图像放大(Image up-scaling)
在CV中,上采样是指从低分辨率变成高分辨率,还原更多图像上的细节
在生信中,上采样是指利用少量的基因表达数据来推测整个基因组的表达模式(从已知的1000个基因的表达数据预测大约20,000个基因的表达水平)

-
数字信号放大:
-
这通常涉及使用插值低通滤波器(例如有限脉冲响应FIR滤波器)来增加信号的采样率。
-
目的是从较低维度的信号中捕捉到更高维度信号的特性。
-
提及了Nyquist率,这是连续信号采样的最小速率,以避免失真。
-
- L1000与RNA-seq对比
- 目标
- 通过测量1000个基因来推断剩余的基因表达。
- 这种方法快速、便宜,且可以应用于数百万种条件。
- 如何选择哪1000个基因进行测量
- 使用“压缩感知”(Compressed Sensing)技术,测量基因的某些组合,从而更好地捕捉到高维数据
- 目标
-
图像放大
-
这与卷积运算的逆运算(去卷积)有关,用于将分辨率较低的图像转换成高分辨率图像。
-
这通常涉及从大量图像中学习的迁移学习。
-
强调了从低维重投影到高维图像的过程。
-
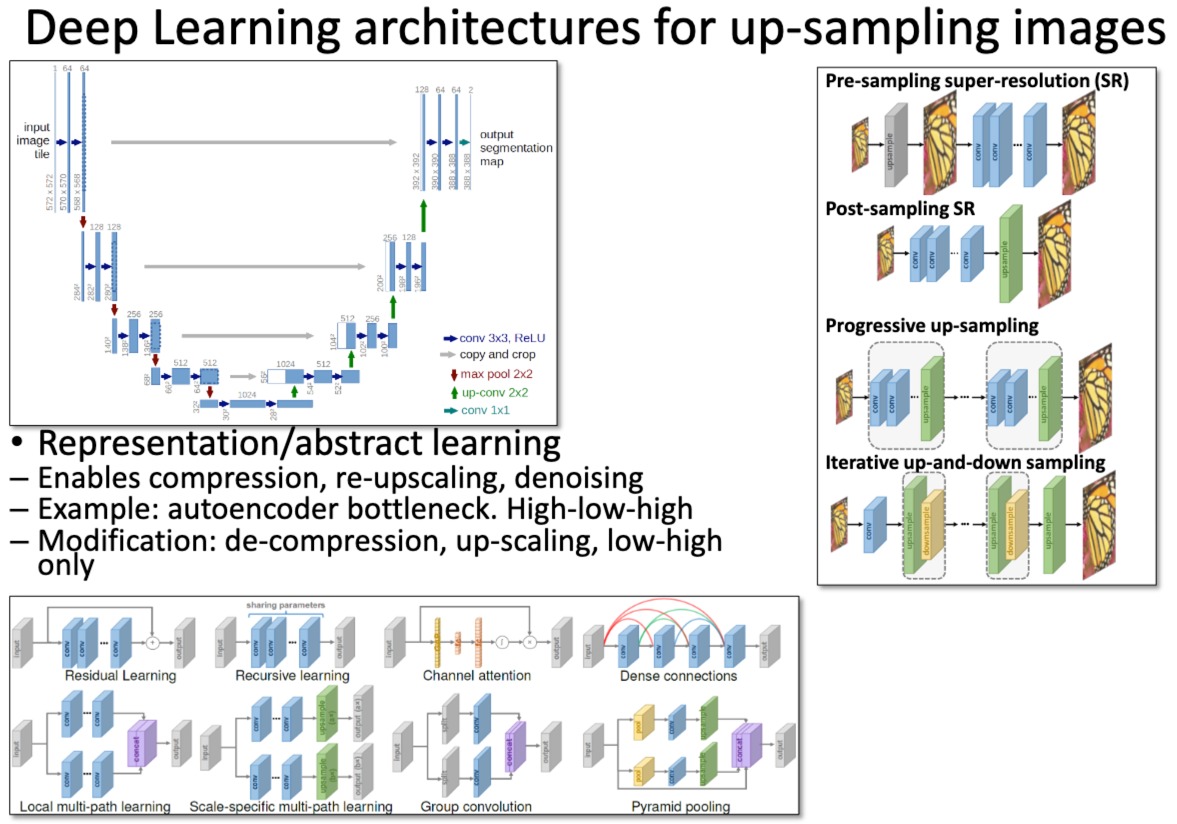
- 多个DL框架,用于增加图像的分辨率
- Representation/abstract learning,让网络学习数据的压缩表示的方法,有利于任务如图像的压缩、去噪和上采样
- 下面有很多模块,都是用于提升网络性能的
- 如残差学习、递归学习、通道注意力、稠密连接等
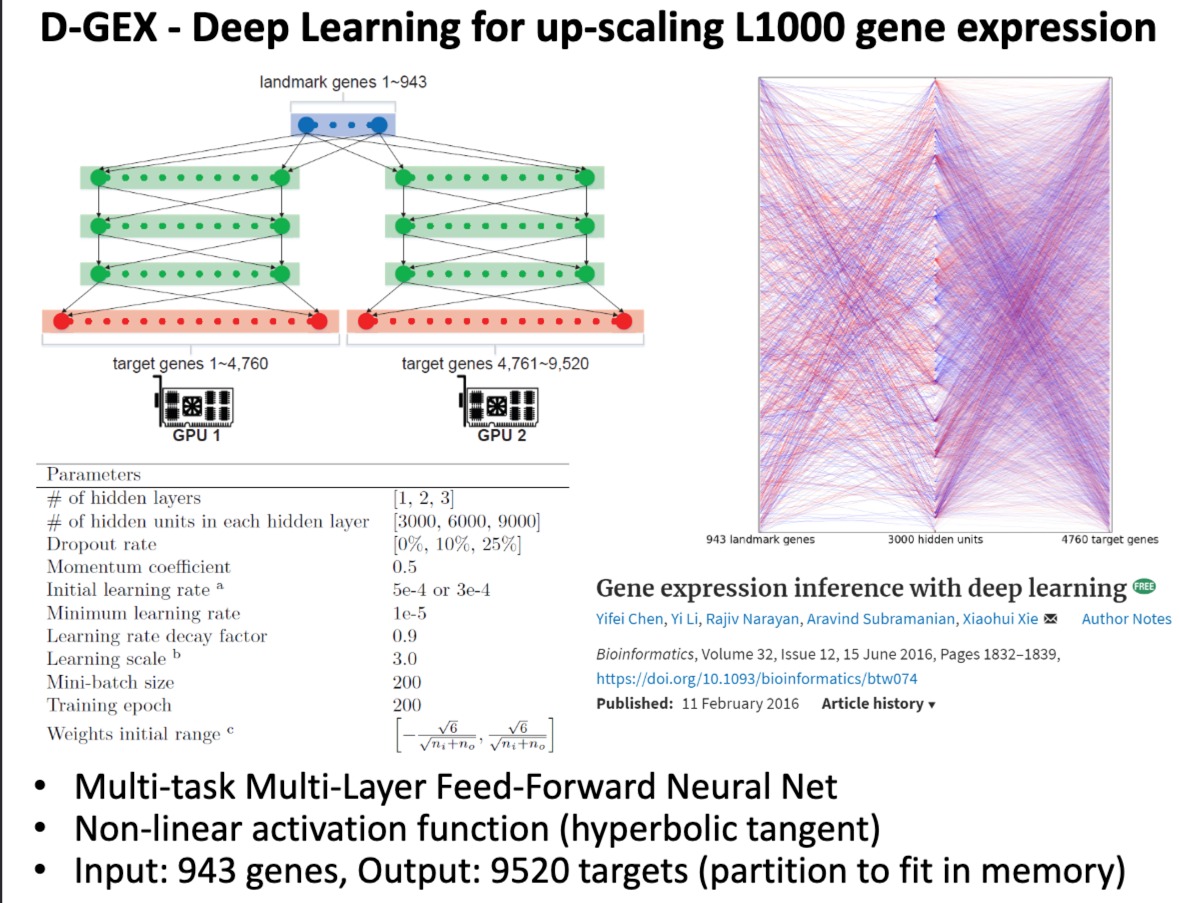
-
一个深度神经网络模型,D-GEX,用来预测基因表达的。
- 一个多任务多层前馈神经网络,使用非线性激活函数(双曲正切函数)
- 输入是943个“landmark”基因的表达数据,输出是预测的9520个目标基因的表达水平
-
不管是在计算机视觉,还是在生物信息
- 深度学习都表现的非常好
3. Compressive sensing
先是介绍了使用随机复合测量(Random Composite Measurements, RCMs)有效生成转录组档案(即基因表达数据)的概念和方法
- 压缩感知(Compressed Sensing
- :这是一种可以从少量观测数据中恢复出完整信号的技术。在这里,它被用来从复合测量中恢复出表达轮廓。
- 随机复合测量(RCMs):这是指用随机的方式组合多个基因的表达数据,以减少必须进行的实际测量数量。
- 推断基因模块活动:通过分析这些随机复合测量,可以推断出不同的基因模块(即一组共同表达的基因)是如何活动的。
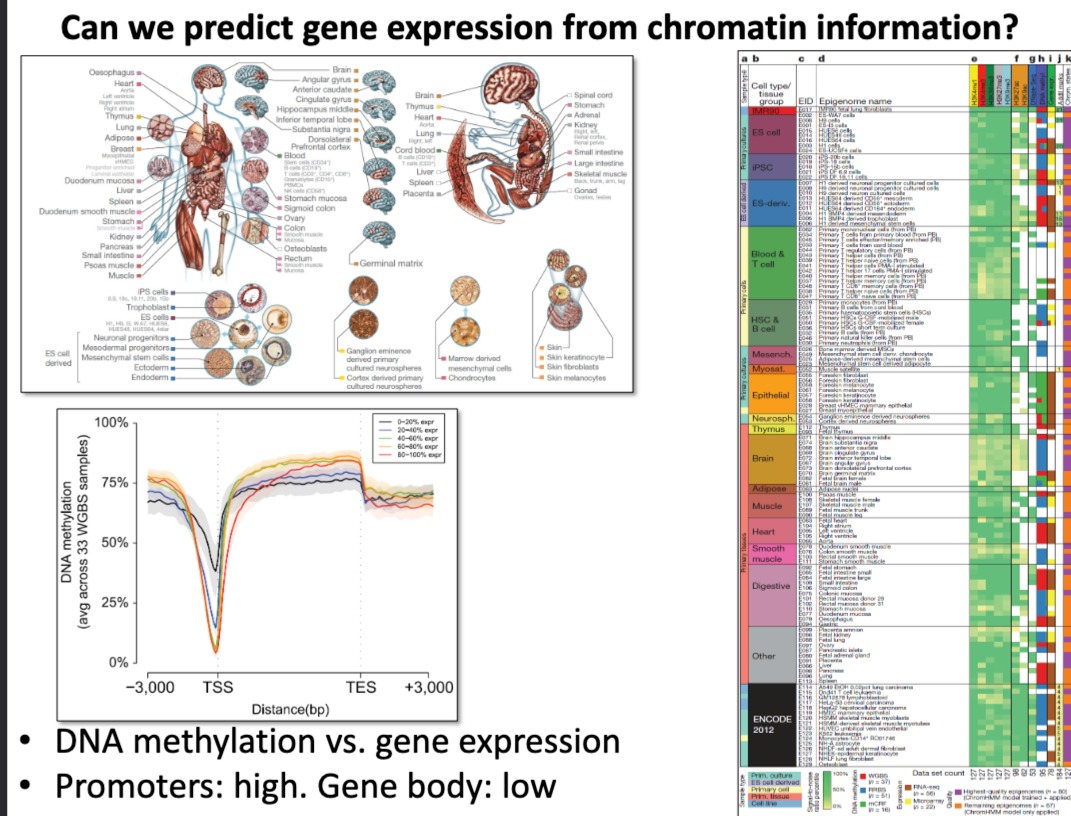
从染色质信息预测基因表达
- 染色质信息包括DNA甲基化、组蛋白修饰等因素,这些都可以影响基因的活性和表达
- DNA甲基化与基因表达
- 折线图展示了从转录起始位点(TSS)到转录终止位点(TES)的DNA甲基化水平,与基因表达水平的相关性。
- 图中,在启动子区域(TSS附近),高甲基化水平与低基因表达水平相关;而在基因体中,甲基化水平则相对较低。
- 右侧热图
- 不同的细胞类型和相关的表观遗传标记
- 每个细胞类型旁边有不同颜色的条形,代表了特定表观遗传特征(如某种特定的组蛋白修饰)的存在或缺失
- 旨在展示如何使用表观遗传学数据(特别是DNA甲基化和组蛋白修饰)来预测不同细胞类型中的基因表达模式。通过分析这些信息,研究人员可以更好地理解基因如何在不同组织和发育阶段被调控。
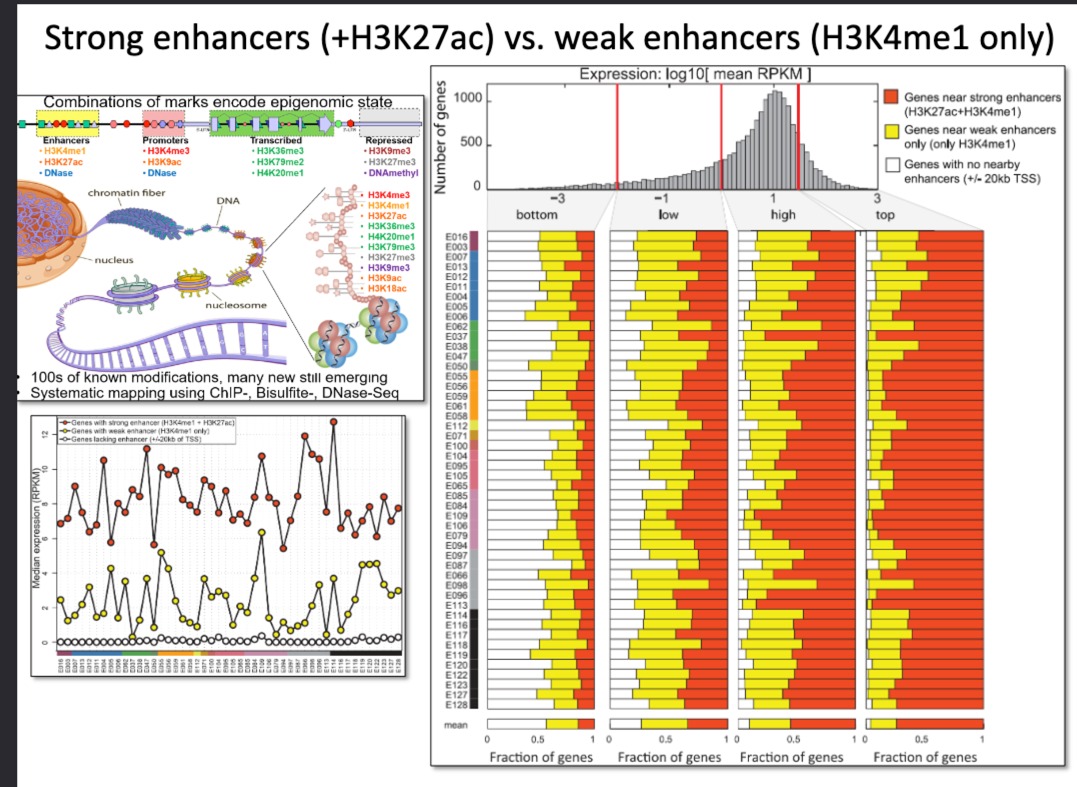
探讨了强增强子(标记为H3K27ac)与弱增强子(仅标记为H3K4me1)在基因表达中的作用
- 左下角:
- 展示了在基因体周围(距离转录起始位点TSS上下约2kb的区域内)的不同表观遗传标记水平的变化。
- 这些数据点代表了有强增强子存在(H3K27ac和H3K4me1均存在)的基因与没有增强子(距离TSS超过20kb)的基因之间的比较。
- 右侧:
- 直方图展示了基因表达的分布,使用对数转换的均值RPKM
- 条形图展示了在基因表达的不同水平(从低到高)下,强增强子和弱增强子附近的基因比例
- 在高表达基因区域周围,强增强子比例越高,类似是这样分析。
- 揭示了不同表观遗传标记在调控基因活性方面的差异作用。可以更好的理解基因表达背后的表观遗传调控机制

这是这节课会听到的第一个客座讲座,后面会详细介绍
3.5 Predicting Reporter Expression from Chromatin Features
尽管我们能够通过比较序列分析、全基因组染色质/转录因子(TF)定位图以及遗传学等方法识别出大量可能的顺式调控元件,但我们仍然不清楚它们的具体功能和调控机制
以下是现代基因调控元件研究的进展的介绍
- “bashing”——传统方法
- 通常涉及将不同的调控元件克隆到报告基因(如荧光素酶或绿色荧光蛋白GFP)前面,并测试它们的活性。
- 缺点
- 生成/克隆个体变体非常耗时
- 酶促/荧光报告器限制了多路复用
- 大规模平行报告基因测定(MPRA
- 测试成千上万的调控元件。MPRA的灵活性允许测试启动子、增强子、沉默子、RNA稳定性元件等。
- 对某些基序(TF结合位点)的破坏是如何影响特定激活子和抑制子的功能的。
- HiDRA(高清晰度报告基因测定):
- HiDRA是一种高定义报告基因测定,具有以下几个关键特点:
- 在单一实验中测试超过700万个片段。
- 不需要合成、大小选择,可以测试长片段。
- 选择可访问的DNA区域,获得高敏感性。
- 3’UTR整合促进自我转录,避免了外源启动子的需要。
- HiDRA是一种高定义报告基因测定,具有以下几个关键特点:
4. Predicting splicing from sequence
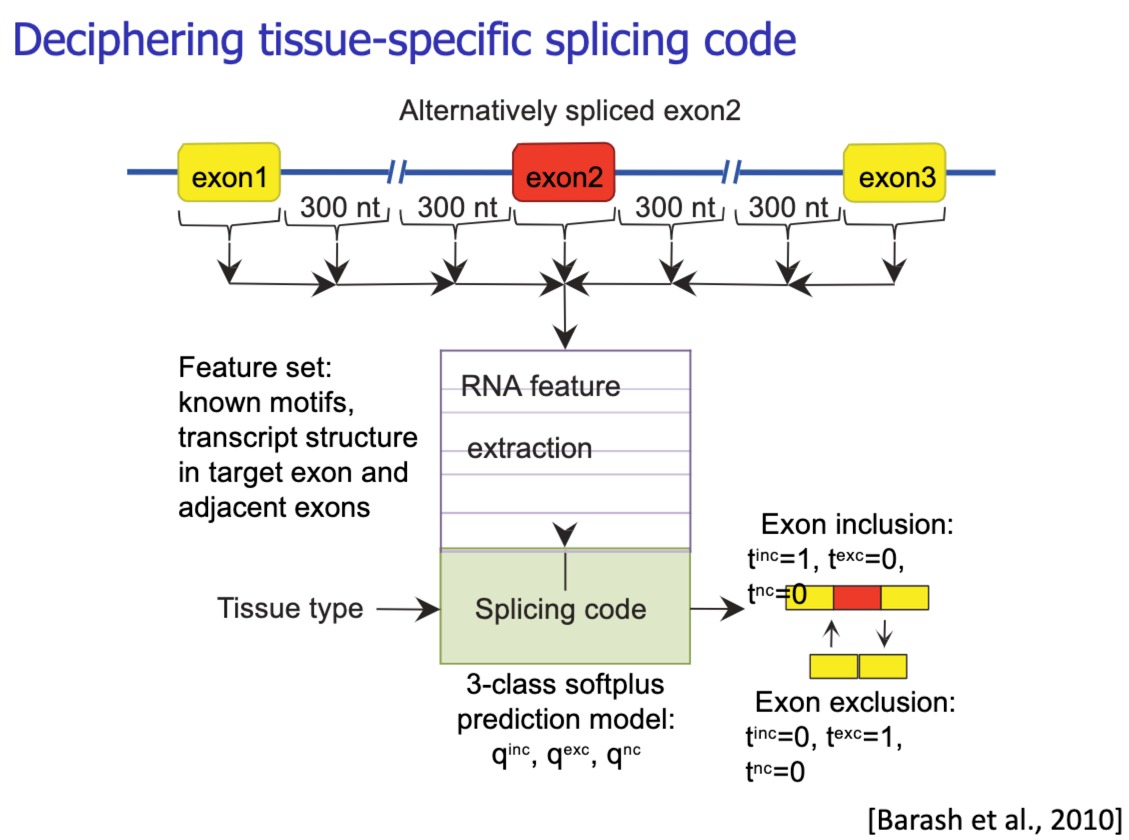
这里解释了如何使用已知的RNA序列特征和剪接代码来预测组织特异性剪接事件。
剪接是基因表达过程中的一个步骤,其中前体mRNA(pre-mRNA)中的内含子被移除,而外显子连接在一起形成成熟的mRNA。
组织特异性剪接是指在特定组织中发生的独特剪接模式。
- 背景:这里三个外显子(exon1, exon2, exon3)。其中exon2是可选剪切,可以包含在成熟mRNA中也可以排除。
- 为了预测剪接,定义了一组特征
- motifs(已知的RNA结合蛋白结合位点)
- 目标外显子
- 相邻外显子的转录结构
- 特征提取
- splicing code是指决定特定外显子是否包含在成熟mRNA中的规则和模式
- 预测模型是一个三分类模型,预测外显子是包括/排除/不确定。
这是第三个客座报告
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!