基因组测序、组装、注释的基本流程
基因组测序:
一代测序——双脱氧终止法(Sanger法)
第一代测序为1977年sanger提出的双脱氧终止法,在理解原理前,必须要知道DNA合成的特点。与RNA合成(转录)不同,DNA合成需要RNA聚合酶先合成一小段RNA片段,为DNA的合成起始提供一个-3’OH(之所以要这样有说法是生物体为了DNA合成起始的保真性),而后续的dNTP延申都借用了上一个dNTP的-3‘OH。Sanger发现,当化学合成出的没有-3’OH的ddNTP(即双脱氧核糖核苷酸,图1)被DNA聚合酶援用延申新链时,由于缺乏3’羟基DNA的合成就会终止掉。天才的
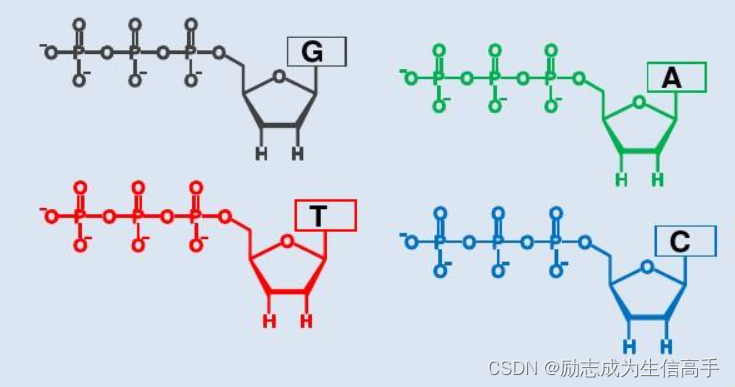
图1
Sanger就想了一个办法,他把四种ddNTP分别加入正常的dNTP中,作为pcr的原料进行pcr。拿ddATP混合试剂举例,由于pcr合成过程中会随机援引正常和不正常的碱基,因此ddATP会在任何对应碱基为T的碱基处被DNA聚合酶援引而停止合成,那么ddATP这一组就会出现各个不同长度的,在各个T处终止的不完全的DNA双链,和未援引ddATP而正常合成的双链。以此类推,那么四组加在一起就会获得每个碱基处都终止的不完全的DNA双链。由于在不同位置终止的链长度都不同,因此其分子量也不同,于是通过聚丙烯酰胺凝胶电泳将其分离就得到不同相对位置的电泳图,那么最轻的,也就是在胶上跑最远的,就是第一个终止的碱基,你当然知道所对应终止的碱基是哪一个,因为被分成了四组(图2),那么从上到下依次读出,就是DNA的序列了。如今的一代测序不需要分成四组这么麻烦,在ddNTP上加上不同的荧光标记就行了。
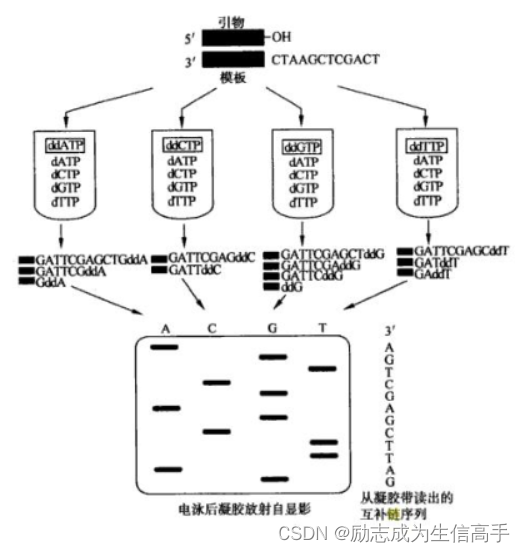
图2
优点:Sanger测序的准确度几乎能达到100%
缺点:通常只能测较小的片段,无法高通量,对于基因组这种庞大的数据量,Sanger法几乎是无力的
二代测序——高通量的illumina
二代测序又叫NGS(next generation sequencing),方法有很多,但是illumina公司几乎是垄断这个市场的,接下来逐步分析illumina基因组测序的步骤和原理
文库(library)制备
将基因打碎成小片段
用规范的方法提取完DNA,质控确保DNA纯度较高后,要把DNA打碎成小片段方便测序,为什么要打碎后面再说。打碎一般采用物理的超声波法,涉及到一些常规的物理知识,超声波在水溶液中会形成小气泡破坏磷酸二酯键,且当共振频率与分子的振动频率一致后也会促使其断裂。超声波主观看来会随机破坏任意一个碱基间的磷酸二酯键,似乎打出来的片段会长短不一,其实不然,只要将超声波的参数控制到位,DNA被打碎到一定长度后就不容易被超声波破坏了。一般打碎的片段长度在200-1000bp不等
对小片段进行前处理(末端齐平,加入接头序列)
需要注意的是,超声波并不破坏链间氢键,尤其是在常温下,dna双链在互补的情况下有比较强的吸引力,但是超声波对磷酸二酯键的破坏参差不齐,甚至会把末端的磷酸基团打成游离的,这样就可能导致某一条链对空,因此需要将双链补齐成平末端。打出来的末端有三种情况(图3):5‘末端突出;3’末端突出;刚好对齐
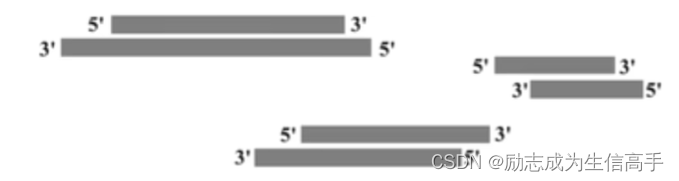
图3
我们知道,DNA的延申是需要3‘OH的,因此处理5’端的突出可以遵循pcr延申的原理,而3‘端的突出一般采取切割,于是选用了即具有延伸功能,又具有3-5外切活性的Klenow酶将所有片段补齐。再用T4激酶为5’端添加一个磷酸基团供后续接头序列的连接。需要注意的是,Klenow酶有和Taq酶相同的功能,即在复制结尾处与模板无关地加上一个A(图4),于是可利用这个特性加上接头序列。
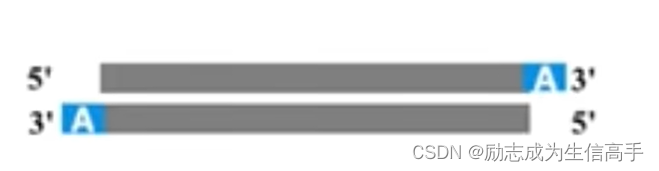
图4
利用T4连接酶,将两个接头序列通过AT互补地连载两端(图5),注意,这里的p5和p7并不是严格互补的,被成为Y字形接头,为什么要这样后面会说。
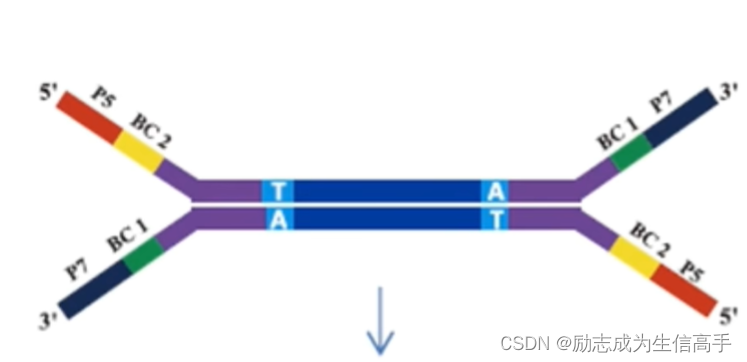
图5
这样一来,整个体系就为:各种长度不等的含接头片段,未被连接上的接头,其他低质量的片段。我们需要的是尽量等长度的含接头片段,如果现在立马加引物跑pcr然后跑胶,那么胶图的结果会像这样(图6),要想获得等长片段,用胶回收会很混乱(可能可以),显然有更好的方法。
图6
磁珠吸附法纯化基因组文库
磁珠是一类将磁性无机粒子与有机高分子结合形成的具有一定磁性的微球。
磁珠的基础结构分为三层,内核是聚苯乙烯材料;中层是磁性物质,一般为氧化铁的混合物;最外层包覆高分子外壳,既避免目标分子直接接触氧化铁,又可以为不同实验提供不同的官能团修饰。本实验中最外层则布满羧基(COO-),DNA分子在盐与PEG混合溶液下会失去表面水化层,暴露磷酸基团,从而分子整体带负电荷,易与正电荷结合,通过溶液中的阳离子与磁珠表面修饰的羧基形成电桥,使得DNA被吸附到磁珠表面(图7)。由于DNA分子带电荷的强度与其长度有关,长度
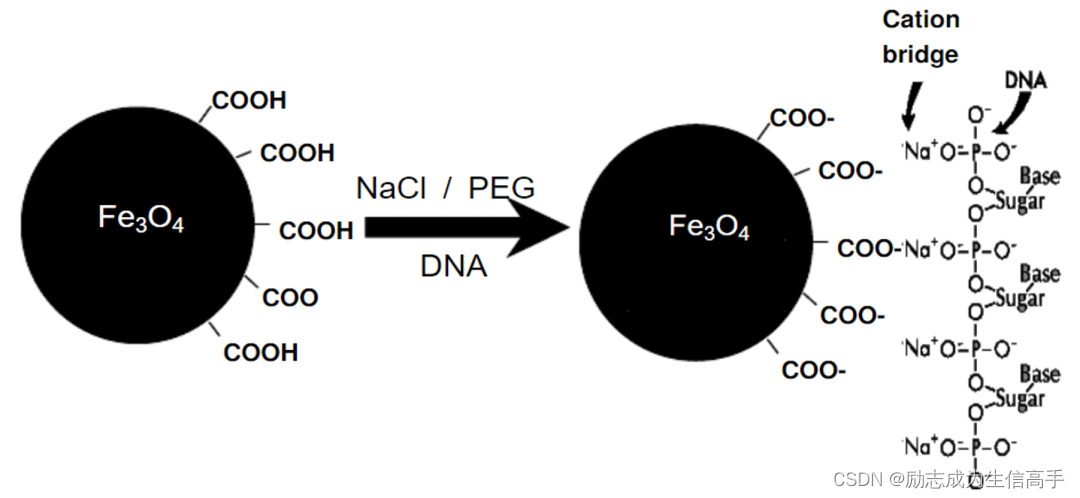 图7
图7
越长,越倾向于在PEG存在条件下发生构象改变,其带的负电荷越大,越易与磁珠结合,因此PEG的浓度是关键因素,通过控制体系成分进行筛选(图8),磁珠将大片段吸附,再用磁力架将大片段沉淀,吸附上清进行第二轮筛选,最终获得的片段经过跑胶确认无误后备用。
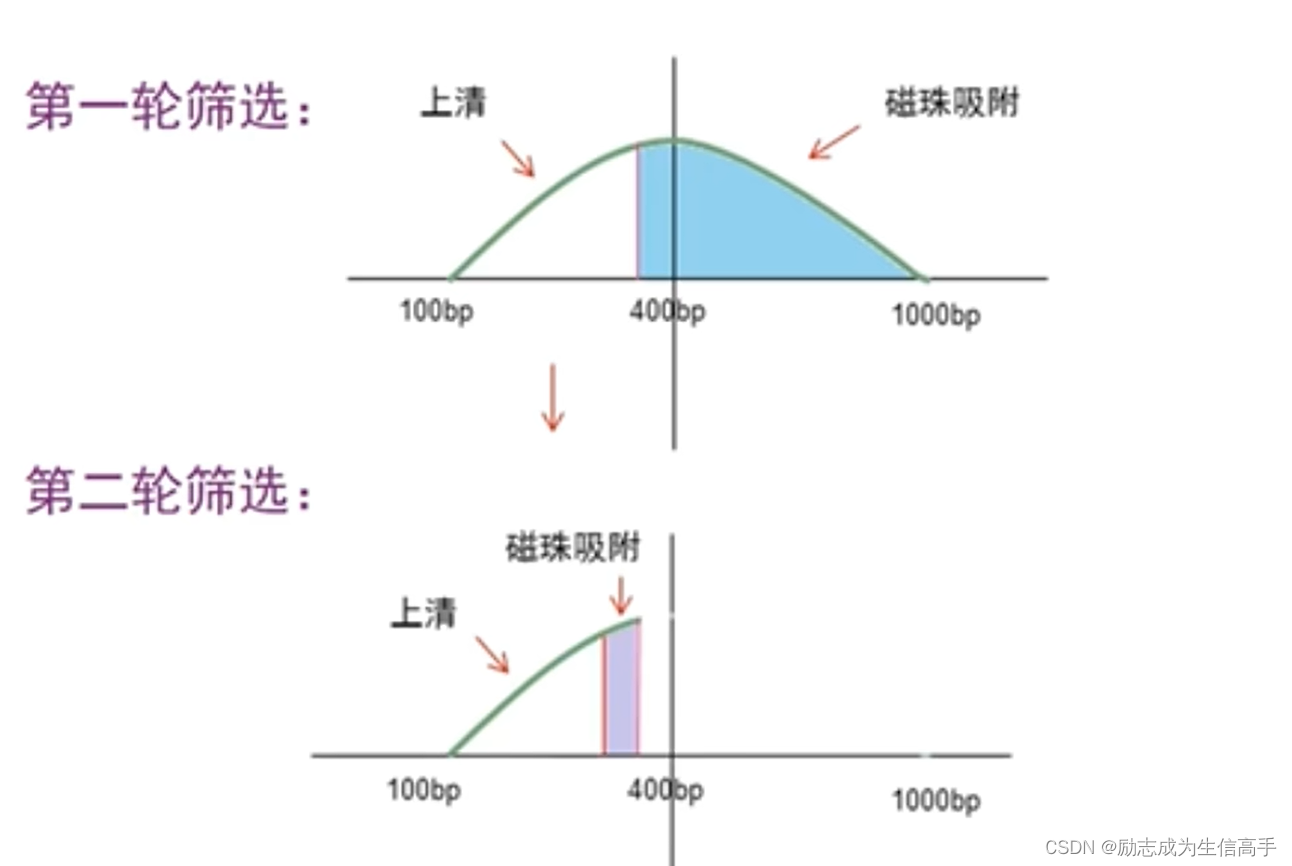 图8
图8
簇生成(桥式pcr)
接头序列结构说明(图9)

图9
P5、P7:与Flowcell结合的序列
index(BarCode):一段有特殊碱基顺序的序列,靠识别这些顺序,可以允许多个测序项目在一次测序中同时进行
RdN SP:真正的测序起始点,后面测序放的引物就与这一段结合
上面三个结构统称为接头序列,注意这三个序列的碱基顺序都是已知的。
将基因组文库与Flowcell连接
用氢氧化钠溶液将图5的双链解开,此刻两条互补链就老死不相往来了,Flowcell内表面(图10)富含与P5和P7分别互补的序列,这些序列与Flowcell内表面以共价键相连

图10
将基因组文库加入Flowcell,这时P5与P5’ 结合,P7与P7’ 结合(图11)。注意,尽管基因组文库的小片段数量有成千上万个,但由于Flowcell面积足够大,其上附着的小接头数量远大于基因组文库小片段的数量,因此每一条被固定的小片段之间间隔是足够大的,通尽常互不影响,管存在特殊情况,即两条不同的序列间隔较近,也会因为高通量的优势弥补这些小问题

终于,迎来了第一次扩增 ,如下所示,左边那条作为模板,固定在槽中的短链作为引物将其复制,由于左边那条尾端没有与槽共价相连,于是用NaOH溶液使双链解旋,再将其洗出Flowcell。

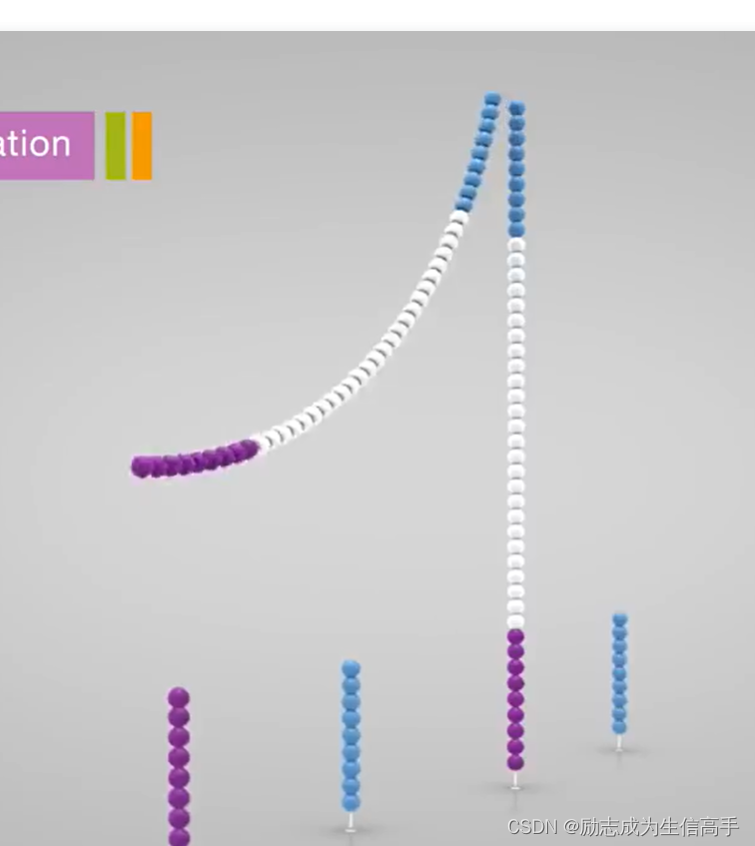
也许你注意到了,图中的片段蓝色那端为什么不与槽内的互补呢?由于体系里仍然有peg与盐溶液,这时的DNA分子结构是偏刚性的,因此是直挺的,待第一次洗脱后,环境内的peg与盐溶液浓度降低,DNA分子就会“疲软”,垂下来与底端的接头连接,并且进行第二次复制。
再加入NaOH溶液使双链解链,便得到两条互补但碱基顺序相反的片段,红笔标注的两个碱基是互补的

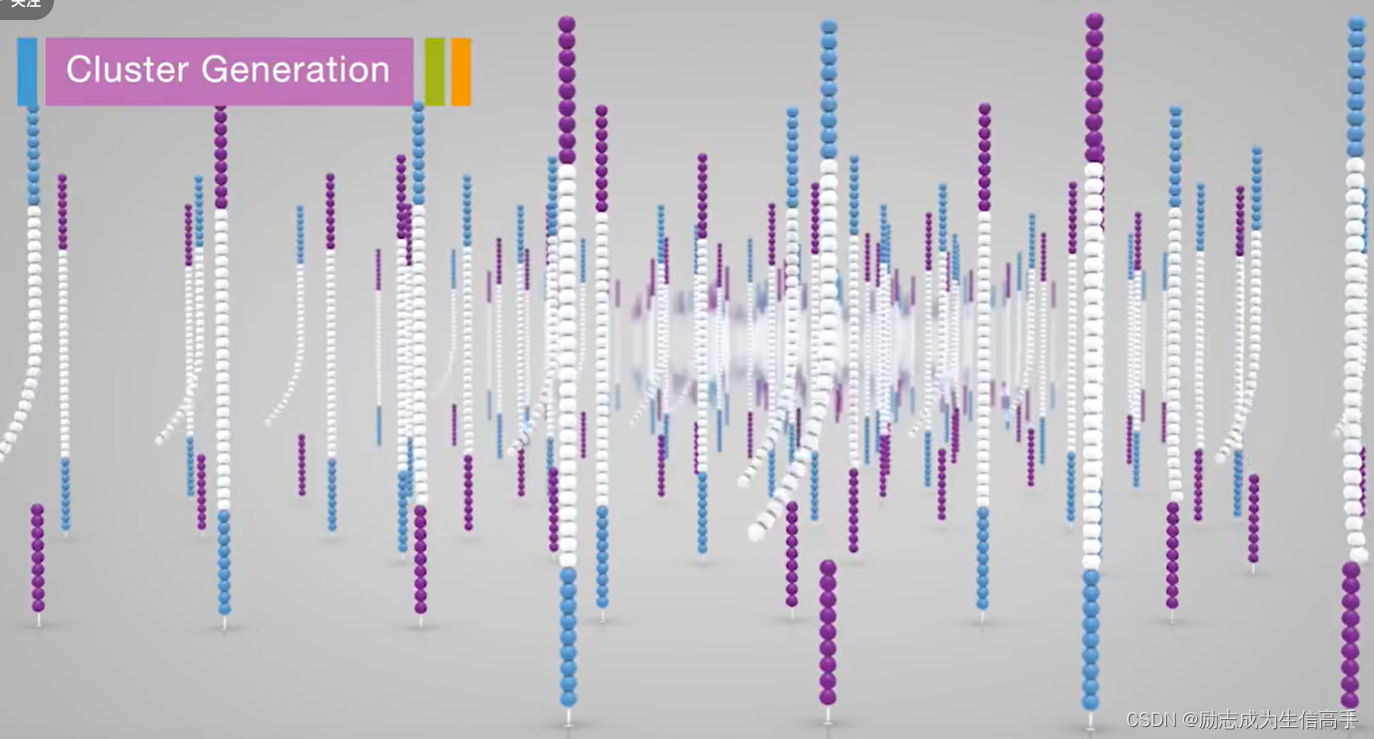
重复这些步骤若干次,便得到数以万计的,上图两条序列的复制产物。上面提到,两条序列不同的片段之间间隔是很远的,因此下图可以认为,相当大一片区域都只是某一个片段的复制产物和与他反向互补的产物。

如果直接测序,上图区域测出的序列会有俩个结果(一个正向的信号,一个互补且反向的信号),由于illumina是捕捉荧光信号的,因此不允许一个区域会有两个不同的信号存在,不然就失去了放大化的初衷,于是用酶将其中一条切掉(单链切割可能我猜也是用限制酶吧,由于接头序列顺序已知,应该会有相关的酶切位点)。如此一来,这个区域就只留下一片序列一模一样的片段了,这样就起到了放大的作用。
 正式开始测序
正式开始测序
可逆阻断终止技术
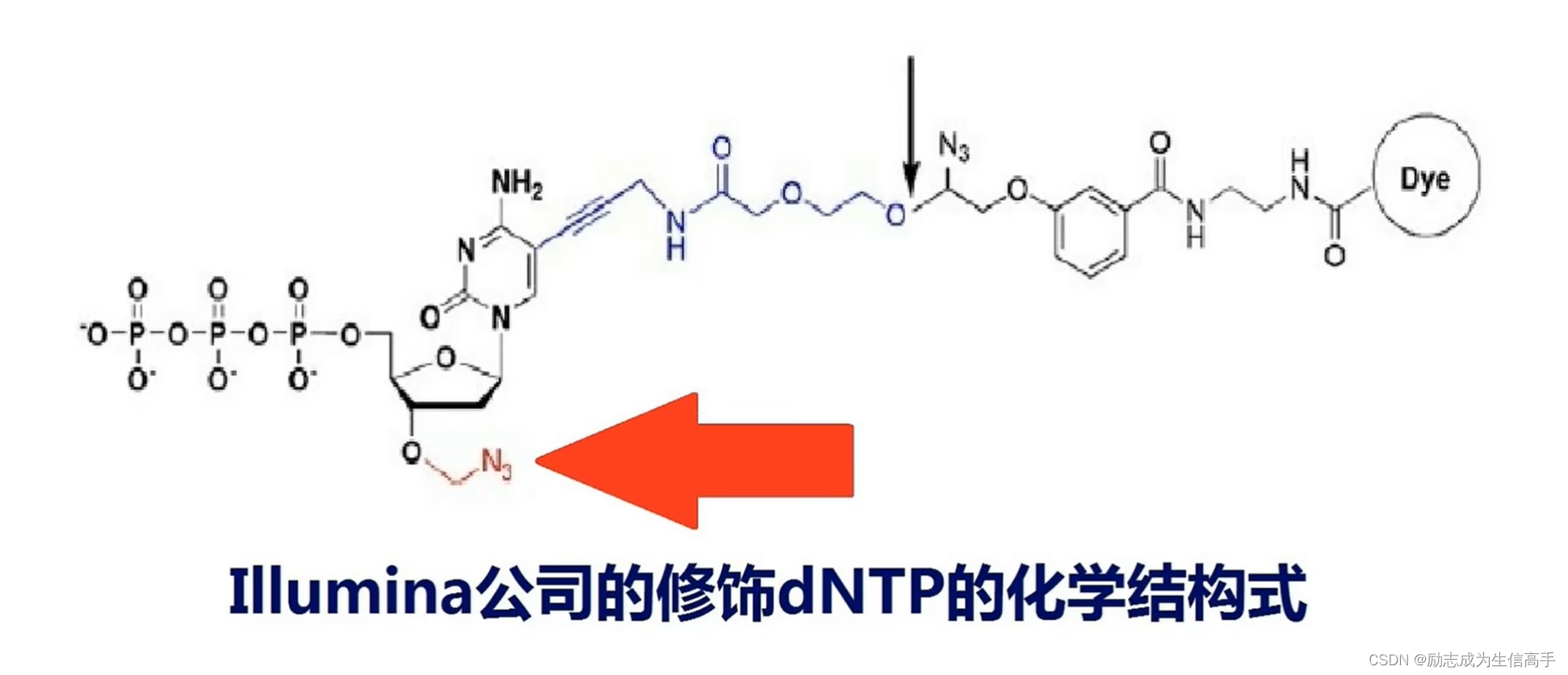
这时illumina测序的核心技术,如图红箭头所指为一个叠氮基团,起到了终止复制的作用,黑箭头所指为一个信号基团,不同碱基带有不同的信号基团。从效果来看,每一轮复制都只能增加一个碱基,扫描捕捉到信号后,再通过切除(包括叠氮基团和荧光基团)和洗脱让其变成一个正常的dNTP继续接收下一个碱基。
测序
添加RdN SP的引物

添加一个碱基——捕捉信号——切除基团+洗脱——迎接下一个碱基

这就是捕捉信号的机子看到的样子,每一个不同颜色的点就代表 图10 那样的“区域”正在测序,这就能解释为什么要桥式pcr,如果如图的每个点都只有1条序列,那么信号是极其难捕捉的。

测序的pcr到哪一步介绍呢?看illumina公司官方视频给出的结果:

看上去是刚好把目的片段测完了,其实这个图比较理想化,事实上你也不知道哪一步刚好把目的片段读完,按我的理解应该是从引物下面的第一个碱基开始测序,一直测到蓝色那一坨,也就是把接头序列也测完为止,由于接头序列都是已知的,而且只会出现在测序结果read的两端,所以后期做数据处理也十分简单。
接头序列有个index序列,是为了可以在一次测序过程中可以测两个样本(比如一个测物种a,一个测物种b),那么如何在如此复杂的测序结果找到哪个是a,哪个是b呢?
用index的引物测一下index的序列就行了!如图,引物下面三个白的碱基就是index,测出来就知道刚才那些信号是属于谁的了,ATCG四种,3个碱基理论上来说一个测序过程能同时给64个样本测序。

有些情况下,另一端接头序列下面也会给一个index,再次经过一个桥式pcr就能知道另一端的index序列是什么了。

理论上来说,测序到现在已经可以结束了,但是为了结果的可靠性,可以选择再进行一次对反向互补链的测序(也就是最开始被酶切掉的反向的序列),用同样的方法,把正向链洗掉,再重复一次测序即可。这里测序的结果是反向互补的,在做数据分析的时候要注意。

数据分析
在拼装基因组时,用正向链和反向链相互补充,知道拼出完整的基因组

每一条蓝的和紫的就是每个测序的结果,被称为read,灰色的就是基因组了

优点:高通量,高速度
缺点:低测序长度,精度没有那么好。之所以要磁珠吸附300bp左右的序列就是因为在测序长于300bp后,荧光信号就会开始混乱,比如某几条在这一轮没有接上碱基,种种原因。有些你拿到手的低质量的序列,会看到很多N,这些就是没测出来的。还有比如在给基因组接在Flowcell时,两个或两个以上的片段在同一个区域里桥式pcr,导致一个区域的信号会混乱
三代测序
笔者承认能力不足,pacbio有点难懂,过几天闲着没事干把它学了
基因组拼装:
陆续更新....
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- nextjs + sharp在 vercel 环境svg转png出现中文乱码
- ARM 作业 按键中断
- Vue3+Ts项目(Naive UI组件)——创建有图标可伸缩的左边菜单栏
- 【PostgreSQL】从零开始:(三十四)数据类型-对象标识符类型
- 开发手机中控软件:从零开始的代码之旅!
- 基于JavaWeb+SSM+Vue校车购票微信小程序的设计与实现系统的设计和实现
- [go] 代理模式
- 回溯法解决n皇后问题(迭代版)
- 一文搞懂Jenkins持续集成解决的是什么问题
- 优化for循环(js的问题)