limma:单通道数据和RNA-seq数据差异性分析标准方法
前言
单通道数据极为流行,三大公司:Affymetrix、Illumina和Agilent的微阵列(microarray)技术产生的很多都是单通道数据。现在的主力的高通量测序机所产生的也是单通道数据,所以只要是被voom标准化(包括了log转化)的RNA-seq数据都可以看作和microarray一样的数据[引用]。这是一个很重要很有用的概念,相信会帮到很多生物信息入门者:
RNA-seq data?+ voom标准化 = microarray data
分析单通道数据其实是最好理解的,和普通的线性回归或者方差分析几乎是一样的。与微阵列数据不同的是,单通道数据通常需要依赖对比,缺少内参,所以R中设计程序的适合没几个标准参数可以用。这一篇文章中,我们仅仅使用最简单的两组对比设计的示例来做最简单的差异性分析。
示例
示例中的样本一共有两组共8个,前4个为正常组织,后4个为肿瘤组织。我们假设我们的表达举证为counts,counts应该长这样:
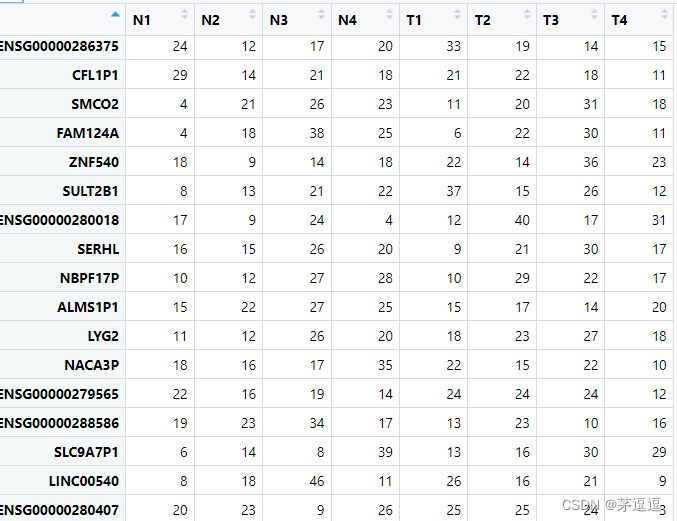 我们需要对这个矩阵进行voom转化,voom就是把 counts 转化为 log2-cpm的过程。
我们需要对这个矩阵进行voom转化,voom就是把 counts 转化为 log2-cpm的过程。
Voom
我们把下列的代码保存,并起名字为voom:
# assume counts is your Expression matrix
#Once a matrix of read counts counts has been created, with rows for genes and columns for samples,
#it is convenient to create a DGEList object using the edgeR package:
dge <- DGEList(counts=counts)
#The next step is to remove rows that consistently have zero or very low counts. One can for
#example use
keep <- filterByExpr(dge, design)
dge <- dge[keep,,keep.lib.sizes=FALSE]
# It is usual to apply scale normalization to RNA-seq read counts, and the TMM normalization
#method in particular has been found to perform well in comparative studies. This can be applied
# to the DGEList object:
dge <- calcNormFactors(dge)
# voom 任选一种:
# 普通:
v <- voom(dge, design, plot=TRUE)
# 直接使用counts数据进行voom
# It is also possible to give a matrix of counts directly to voom without TMM normalization, by
v <- voom(counts, design, plot=TRUE)
# 如果你觉得原始数据噪声很大,也可以
#If the data are very noisy, one can apply the same between-array normalization methods as would
#be used for microarrays, for example:
v <- voom(counts, design, plot=TRUE, normalize="quantile")
设置分组
接下来就是设计矩阵了,根据要不要"对比矩阵",我们得到type分配了分组(处理):
type <- c("normal","normal","normal","normal","tumor","tumor","tumor","tumor")设计矩阵分两种情况讨论
第一种:不要设计矩阵
# Here the first coefficient estimates the mean log-expression for wild type mice and plays the role
# of an intercept.?
Group <- factor(type, levels=c("normal","tumor"))
design <- model.matrix(~Group)
colnames(design) <- c("normal","tumorvsnormal")
design
source("voom")
fit <- lmFit(v, design)
fit <- eBayes(fit)
topTable(fit, coef="tumorvsnormal", adjust="BH")
?第二种:要设计矩阵
#第二种设计
#The second coefficient estimates the difference between mutant and wild type.
#Differentially expressed genes can be found by
design <- model.matrix(~0+type)
colnames(design) <- c("normal","tumor")
source("voom")
fit <- lmFit(v, design)
cont.matrix <- makeContrasts(tumorvsnormal=tumor-normal, levels=design)
fit2 <- contrasts.fit(fit, cont.matrix)
fit2 <- eBayes(fit2)
topTable(fit2, adjust="BH")结果
我们看一下结果:
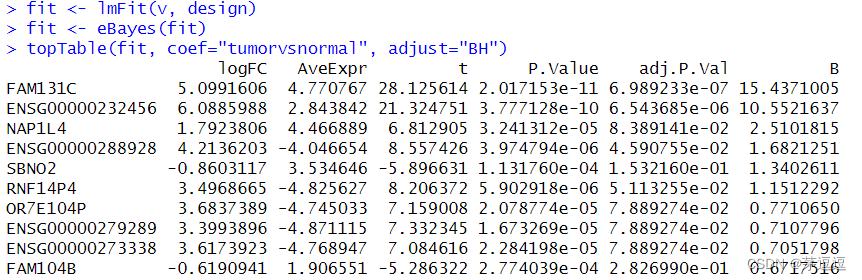 我们来看结果
我们来看结果
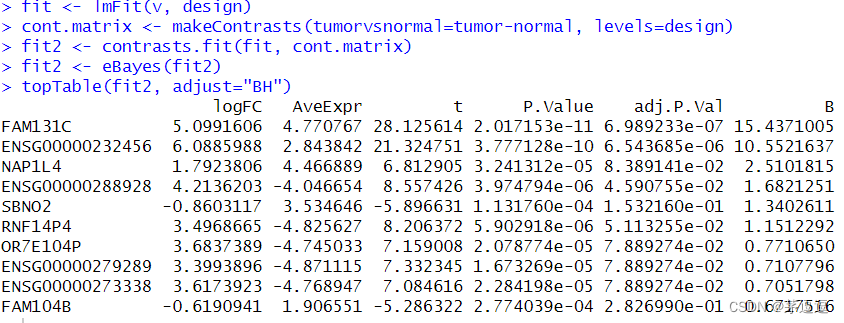
你会发现结果是一样的,以上是针对单通道数据和voom标化 RNA-seq数据而言的二组对比,也是最常用的差异性分析方法,分享给大家。?
引用
Law, C.W., Chen, Y., Shi, W., and Smyth, G.K. (2014). Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biology 15, R29
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- C语言中关于指针的理解
- 智能优化算法应用:基于人工蜂鸟算法3D无线传感器网络(WSN)覆盖优化 - 附代码
- wsl相关
- IOS工程师,再不转型!失业在所难免!
- 利用深度学习图像识别技术实现教室人数识别
- 获取税率GET_TAX_PERCENTAGE
- 微信小程序:布局样式
- qiankun微前端原理及部分源码解读
- 【C语言】动态内存管理之4个内存函数`malloc`,`free`,`calloc`和`realloc`深度了解
- LeetCode刷题笔记