知识引导的分子生成扩散模型 - KGDiff 评测
一、背景介绍
KGDiff模型是一个基于口袋的知识引导的3D分子生成的扩散模型,来源于上海交通大学计算机学院涂仕奎教授的文章:
《KGDiff: towards explainable target-aware molecule generation with knowledge guidance》。文章链接:*KGDiff: towards explainable target-aware molecule generation with knowledge guidance (silverchair.com)。该文章发表在《Briefings in Bioinformatics》期刊上。

基于口袋的分子生成模型之前有介绍过targetdiff,FLAG等。其中,KGDiff与TargetDiff类似,KGDiff模型也是一个扩散模型,应针对的是口袋条件下的3D分子生成。KGDiff的创新点在于:KGDiff模型利用领域知识,例如,vina score,指引分子生成过程中的去噪过程,可生成高结合力的分子。此外,KGDiff还是一个原子层级可解释性的模型,在生成分子时,同时给出生成分子预测score,原子层面的score。
二、模型介绍
KGDiff的模型结构如下图:
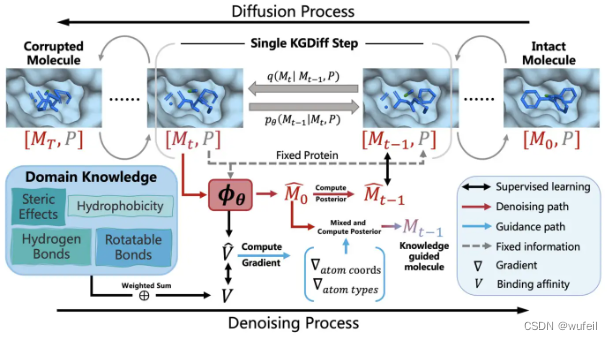
在扩散过程中,分子在 T 个时间步长内逐渐添加预定义的噪声。模型 φ(θ) 被训练为从噪声分子中重建分子,并预测完整分子 M0 和固定蛋白质 P 之间跨反向 T 步骤的结合亲和力 v,即在每一步去噪过程中,神经网络除了预测上一步分子中原子的位置还有原子类型以外,还要预测蛋白和分子的结合力 V;在模型中,蛋白口袋P事固定的,即其嵌入向量不会被更新。生成分子时,进一步利用预测的 V 来引导去噪过程实现高结合亲和力,通过梯度更新分子的坐标和节点类型。 在整个过程中保持蛋白质结构固定。
因此,KGDiff与TargetDiff的区别是:
KGDiff的神经网络?φ(θ),除了要预测去噪后分子原子的坐标与类型,还要预测分子与口袋的结合力,因此,此时的神经网络不单单是一个去噪网络,还是领域知识的专家网络。TargetDiff的神经网络仅仅预测去噪后原子的坐标和类型。此外,在分子生成过程中,KGDiff会使用预测的分子与口袋的结合力的梯度(结合力对原子坐标X和原子类型H的梯度)更新原子坐标和原子类型,以实现生成的分子具有更高亲和力。当然,模型还可以简单的改装为clogP等。这也就是为什么KGDiff是一个knowledge guidance知识引导的扩散模型。
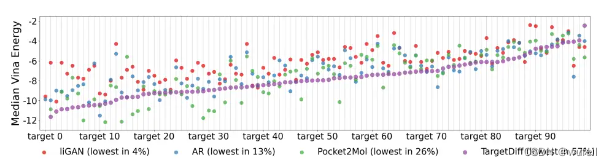
传统基于口袋的分子生成扩散模型,往往仅仅是从数据角度拟合分子和蛋白质之间的联合分布,而忽略了它们之间的结合亲和力,因此生成分子与口袋的结合力并不是很好,体现在生成分子的vina score均值与参考分子vina score相近。如下图(来源于TargetDiff文章):
三、模型性能
3.1 专家网络性能
KGDiff的神经网络?φ(θ)能否预测噪音状态下分子与口袋的结合力是该方法能否生成高亲和力分子的关键。作者给出神经网络?φ(θ)对亲和力预测的结果,如下图:
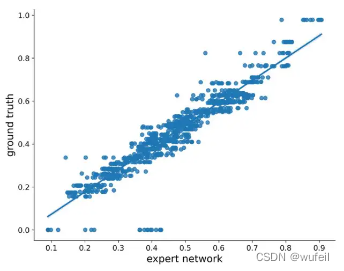
上图中,作者比较了含噪音状态下(每个10个step),专家网络预测值与真实vina score之间的相关性。皮尔森相关系数为0.94,说明专家网络了可以从含噪音的分子中学习到vina score,这也奠定了梯度引导的基础。
3.2 生成分子的QED,SA 以及 Vina score
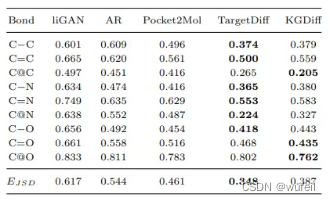
作者对比了liGAN, GraphBP, AR, Pocket2Mol, TargetDiff与KGDiff模型。结果见下表:
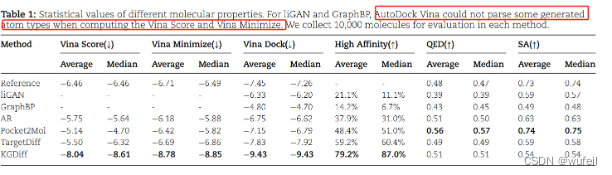
作者分别比较了 生成的3D分子与口袋的结合力打分,即vina score生成的3D分子与口袋进行局部最小化以后的结合力打分,即Vina Minimize,以及生成的3D分子经过重对接(全局优化,构象生成,位置采样,结合力打分)后的打分,即Vina Dock。不管是原味打分下的vina score,局部优化的Vina Minimize,还是全局优化的Vina Dock, KGDiff的打分都会优于其他模型。
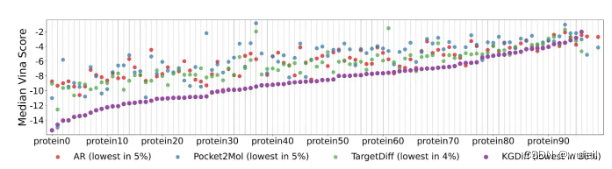
各同体系下,vina score的对比见下图。可以看出,KGDiff生成的分子的vina score, 远低于TargetDiff等模型,效果非常明显。说明生成分子的结合力更强。
3.3 案例研究-可解释性
对于可解释性,作者给了三个例子。为了分析分子生成的结果,作者将生成的分子及其目标蛋白可视化,并将分子中每个原子的打分机型0-100的归一化,见下图。

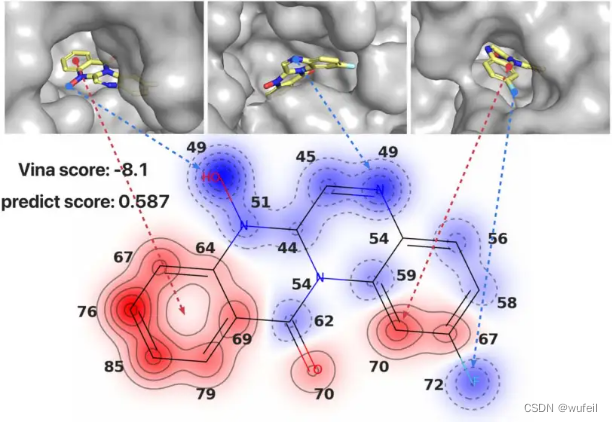

作者定义了pocket hole interface,即指蛋白质口袋表面与外界环境相连的区域。大多数得分较低的原子通常位于“pocket hole interface”附近,并且自身周围的蛋白质原子数量较少。
3.4 不同蛋白的案例
在GPCR, Kinase等靶点上,KGDiff也展示出了类似的结果,见下图。效果仍然是非常明显,对比参考分子的vina score等打分明显下降。

3.5 其他测试
作者工作非常丰富,除了上述的案列测试。还有大量关于影响梯度引导对生成分子因素的测试,包括:vina socre与口袋提及的关系,见下图:

生成分子中原子打分与周围蛋白原子数量之间的关系,如下图:

这两个结果都说明,KGDiff更倾向于大口袋,在口袋内部生长分子,以达到提高蛋白-分子结合力的目标。
此外,作者对同源的体系(TNKS1和 TNKS2)生成的分子,进行了测试,比较两个体系生成分子交叉对接的结果,以及交叉相似性的结果。结果如下表。结果说明,KGDiff生成的分子具有一定的口袋亚型选择性。


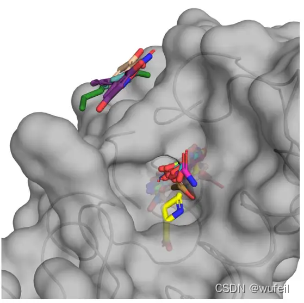
此外, 作者还测试了平坦口袋生成分子的效果,如下图。平坦口袋时,可能是生长位置识别出错,导致两边生长的情况。
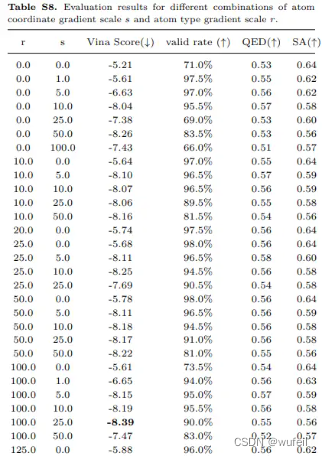
作者还研究了,不同坐标和原子类型的引导强度,会对生成分子结合力的影响,如下图:
同时,作者测试了不同去噪步数下,预测vina score的变化情况,可以看出,去噪步数>100以后,vina score基本稳定,如下图。

还有就是关于生成分子价键长度的统计与比较。
四、模型测评
3.1?环境安装
创建环境,安装pytorch,scipy, numpy等
conda create -n KGDiff python=3.9
conda activate KGDiff
conda install pytorch==1.11.0 torchvision==0.12.0 torchaudio==0.11.0 -c pytorch
conda install pytorch-scatter pytorch-cluster pytorch-sparse==0.6.13 pyg==2.0.4 -c pyg
pip install pyyaml easydict lmdb
pip install numpy==1.21.6 pandas==1.4.1 tensorboard==2.9.0 seaborn==0.11.2
pip install Pillow==9.0.1
pip install scipy==1.7.3
conda install -c conda-forge openbabel
pip install meeko==0.1.dev3 vina==1.2.2 pdb2pqr rdkit安装autodocktools,Linux版本
# install autodocktools
# for linux
python -m pip install git+https://github.com/Valdes-Tresanco-MS/AutoDockTools_py3
安装autodocktools,Windows版本
# install autodocktools
# for windows
python.exe -m pip install git+https://github.com/Valdes-Tresanco-MS/AutoDockTools_py3复制项目代码
git clone https://github.com/CMACH508/KGDiff.git复制完成后,目录如下:
.
├── configs
│ ├── sampling.yml
│ └── training.yml
├── datasets
│ ├── __init__.py
│ ├── pl_data.py
│ └── pl_pair_dataset.py
├── LICENSE
├── MANIFEST.in
├── models
│ ├── common.py
│ ├── molopt_score_model.py
│ └── uni_transformer.py
├── README.md
├── reproduction.ipynb
├── scripts
│ ├── cross_dock.py
│ ├── data_preparation
│ │ ├── clean_crossdocked.py
│ │ ├── extend_protein_extract.py
│ │ ├── extract_pockets.py
│ │ ├── __init__.py
│ │ ├── inject_affinity.py
│ │ └── split_pl_dataset.py
│ ├── dock_extended_baseline.py
│ ├── evaluate_diffusion.py
│ ├── evalueate_valnet.py
│ ├── __init__.py
│ ├── sample_diffusion.py
│ ├── sample_for_pocket.py
│ └── train_diffusion.py
├── setup.py
└── utils
├── data.py
├── evaluation
│ ├── analyze.py
│ ├── atom_num_config.py
│ ├── atom_num.py
│ ├── docking_qvina.py
│ ├── docking_vina.py
│ ├── eval_atom_type.py
│ ├── eval_bond_length_config.py
│ ├── eval_bond_length.py
│ ├── fpscores.pkl.gz
│ ├── sascorer.py
│ ├── scoring_func.py
│ └── similarity.py
├── misc.py
├── reconstruct.py
├── train.py
├── transforms.py
├── vina_rules.py
├── visualize.py
└── warmup.py
7 directories, 47 files4.2 下载训练数据集
数据下载链接:KGDiff
其中,data.zip是数据集,logs_diffusion.zip为模型的checkpoint。将下载的zip文件放置在./项目主目录下,然后均解压。
解压数据集
unzip data.zip
unzip benchmark.zip
unzip logs_diffusion.zip
unzip misc_results.zip注:在解压data.zip的时候,可能会出现以下报错:
error: invalid zip file with overlapped components (possible zip bomb)
To unzip the file anyway, rerun the command with UNZIP_DISABLE_ZIPBOMB_DETECTION=TRUE environmnent variable此时,在~/.bashrc文件中添加如下内容,然后 bash ~/.bashrc即可正常解压。
UNZIP_DISABLE_ZIPBOMB_DETECTION=TRUE
export UNZIP_DISABLE_ZIPBOMB_DETECTION注:我们下载完成的文件,是经过处理以后的,里面已经包含了vina score。
4.3 复现分子生成部分代码
(1)针对CrossDocked2020验证集中的第一个体系,采样
python scripts/sample_diffusion.py \
--config ./configs/sampling.yml \
-i 0 \
--guide_mode joint \
--type_grad_weight 100 \
--pos_grad_weight 25 \
--result_path ./cd2020_pro_0_res运行输出:
但是,运行过程中遇到报错:
Traceback (most recent call last):
File "./KGDiff-main/scripts/sample_diffusion.py", line 259, in <module>
main()
File "./KGDiff-main/scripts/sample_diffusion.py", line 230, in main
pred_pos, pred_v, pred_exp, pred_pos_traj, pred_v_traj, pred_exp_traj, pred_v0_traj, pred_vt_traj, pred_exp_atom_traj, time_list = sample_diffusion_ligand(
File "./KGDiff-main/scripts/sample_diffusion.py", line 134, in sample_diffusion_ligand
all_pred_exp_traj = torch.stack(all_pred_exp_traj,dim=0).numpy()
RuntimeError: stack expects each tensor to be equal size, but got [4] at entry 0 and [2] at entry 2000(2)针对PDBBind2020验证集 中的第一个体系,采样:
更改--guide_mode joint参数为--guide_mode pdbbind_random,让模型针对PDBBind2020的第一个体系采样分子,也发生一样的报错。
(3)对文章中S4表格(如下图)的蛋白,进行分:子生成。尝试按照,github中的如下命令:
python scripts/sample_for_pocket.py \
--pdb_idx 0 \
--protein_root ./data/extended_poc_proteins/ \
--guide_mode joint \
--type_grad_weight 100 \
--pos_grad_weight 25 \
--result_path ./extended_pro_0_res出现报./data/extended_poc_proteins/index.pkl文件缺失错误:
File "./KGDiff-main/scripts/sample_for_pocket.py", line 102, in main
with open(os.path.join(args.protein_root, 'index.pkl'), 'rb') as f:
FileNotFoundError: [Errno 2] No such file or directory: './data/extended_poc_proteins/index.pkl'此时,需要解压./data目录下的extended_poc_proteins.zip文件,可以解决这个错误。
再次运行,则出现如下与之前类似的错误:
File "./KGDiff-main/scripts/sample_diffusion.py", line 134, in sample_diffusion_ligand
all_pred_exp_traj = torch.stack(all_pred_exp_traj,dim=0).numpy()
RuntimeError: stack expects each tensor to be equal size, but got [4] at entry 0 and [2] at entry 2000至此,作者提供的github中的分子生成方式,均指向同一错误。此处错误,简单处理后,即可正常运行。
解决该问题后,可以运行分子生成。
另外,在查看了KGDiff的代码后,发现最后生成的分子会被torch保存成pt文件,而不是我们熟悉的sdf或者xyz文件。生成分子的文件目录为:
.
├── log.txt
├── result_8a1z_C_KVU_pocket10_0.pt
└── sample.yml
0 directories, 3 fileslog.txt为log日志文件,pt文件为生成分子,sample为生成分子使用的参数。
生成100个分子,大约花费1个小时10分钟,生成效率挺慢的。
4.4 评估生成的分子
python scripts/evaluate_diffusion.py \
--sample_path ./extended_pro_0_res \
--protein_root ./data/extended_poc_proteins/在对evaluate_diffusion.py 修改以后,可以产生部分重构成功的分子,保存成sdf格式(保存了生成分子的构象,未经优化)。结果见下图:
从生成的分子的有效性上看,KGDiff生成分子的有效率非常的低,存在大量断裂的分子,存在离散的原子类型。有可能是作者提供的checkpoint训练程度不够,也有可能模型的性能就是如此。更有可能是,能达到那么强结合力的分子数量本来就很少,少于100个,KGDiff模型在不断尝试,尽量给出正确的分子。
同时我们保存相应的smiles,结果中的几个例子如下:
C=C(O)CC.C=CC(=C)C(CC)C(CCC)CCCCC(=O)C(N)=O
C=CC.C=CCC.C=CCCCCC1CC(C(N)C(=O)F)CC1O.COCO.F.O.O
C=CC1CCC2CC(C(O)C(O)CCC(O)(CC(=C)PC(C)=O)C(O)CC)C12
CC1(C2CC=CCCCCC2)C(O)C(O)(C(=O)C=O)C1C(=O)O
C.C#C.C=CCOC.CC(O)(O)C(=O)O.CC1=CCC(COCC2=CC=NC3CNCCC23)=CC1.CC=CCC(O)(O)C(O)CC(N)=O.CCCC(=O)O.CCO.Cc1ccc(O)cc1
C=C(C(C)CCCC(C)C)C(F)CF.CC(O)=C=CC=CC1=NCCC1.F.O
C=CCC(CPPC1(C)OC2CC(=N)C21)C(=C)CC(=CO)CCCC.CC=O.F
C#CC1CC2(C)CC(C)(C3CCCC3)CCCCC12.CNC(=O)C(=O)C(O)C(O)CF
N=CC1=CCC(NOCCN=CC(O)C2C(=O)OC(C3CCC=CC3N)C(=O)C2O)=CC1从中可以看出生成分子中存在大量分子片段。
另一方面,也注意到作者并不是通过xyz以及原子类型,使用obabel软件生成分子的sdf文件,而是使用了XXX函数,这个函数在diffusion相关的模型中比较少见。这也可能是导致生成分子有效率低的原因。
非常可惜,作者的文章中并没有提供关于分子有效率的数据,所以,并不清楚这里面的有效率是否与作者的结果相符合。
4.5 复现训练模型部分代码
尝试按照github的方法训练模型
python scripts/train_diffusion.py \
--config configs/training.yml会遇上以下错误:
TypeError: Descriptors cannot be created directly.
If this call came from a _pb2.py file, your generated code is out of date and must be regenerated with protoc >= 3.19.0.
If you cannot immediately regenerate your protos, some other possible workarounds are:
1. Downgrade the protobuf package to 3.20.x or lower.
2. Set PROTOCOL_BUFFERS_PYTHON_IMPLEMENTATION=python (but this will use pure-Python parsing and will be much slower).此时,升级tensorboard即可。
pip install tensorboard -U再次运行
python scripts/train_diffusion.py \
--config configs/training.yml输出:
[2024-01-03 21:22:47,440::train::INFO] Namespace(config='configs/training.yml', device='cuda', logdir='./logs_diffusion', ckpt='', tag='', value_only=False, train_report_iter=200)
[2024-01-03 21:22:47,440::train::INFO] {'data': {'name': 'pl', 'path': './data/crossdocked_v1.1_rmsd1.0_pocket10', 'split': './data/crossdocked_pocket10_pose_split.pt', 'transform': {'ligand_atom_mode': 'add_aromatic', 'random_rot': False}}, 'model': {'model_mean_type': 'C0', 'beta_schedule': 'sigmoid', 'beta_start': 1e-07, 'beta_end': 0.002, 'v_beta_schedule': 'cosine', 'v_beta_s': 0.01, 'num_diffusion_timesteps': 1000, 'loss_v_weight': 100.0, 'loss_exp_weight': 1.0, 'sample_time_method': 'symmetric', 'affinity_with_diffusion': False, 'use_classifier_guide': True, 'time_emb_dim': 0, 'time_emb_mode': 'simple', 'center_pos_mode': 'protein', 'node_indicator': True, 'model_type': 'uni_o2', 'num_blocks': 1, 'num_layers': 9, 'hidden_dim': 128, 'n_heads': 16, 'edge_feat_dim': 4, 'num_r_gaussian': 20, 'knn': 32, 'num_node_types': 8, 'act_fn': 'relu', 'norm': True, 'cutoff_mode': 'knn', 'ew_net_type': 'global', 'num_x2h': 1, 'num_h2x': 1, 'r_max': 10.0, 'x2h_out_fc': False, 'sync_twoup': False, 'pred_exp_from_all': False}, 'train': {'seed': 2021, 'batch_size': 4, 'num_workers': 1, 'n_acc_batch': 1, 'max_iters': 10000000, 'val_freq': 1000, 'pos_noise_std': 0.1, 'max_grad_norm': 8.0, 'bond_loss_weight': 1.0, 'optimizer': {'type': 'adam', 'lr': 0.001, 'weight_decay': 0, 'beta1': 0.95, 'beta2': 0.999}, 'scheduler': {'type': 'plateau', 'factor': 0.95, 'patience': 15, 'min_lr': 1e-05}}}
[2024-01-03 21:22:47,440::train::INFO] Loading dataset...
[2024-01-03 21:22:47,446::train::INFO] Training: 99990 Validation: 100 Test: 0
[2024-01-03 21:22:47,446::train::INFO] Building model...
[2024-01-03 21:22:49,121::train::INFO] protein feature dim: 27 ligand feature dim: 13
[2024-01-03 21:22:49,122::train::INFO] # trainable parameters: 2.8413 M
[2024-01-03 21:22:49,789::train::INFO] [Train] Iter 0 | Loss 1.590827 (pos 1.234858 | v 0.003544 | exp 0.001567) | Lr: 0.001000 | Grad Norm: 5.512002
Validate: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 25/25 [00:07<00:00, 3.53it/s]
Traceback (most recent call last):之后,会遇到如下错误:
Traceback (most recent call last):
File "/home/KGDiff-main/scripts/train_diffusion.py", line 371, in <module>
main()
File "/home/KGDiff-main/scripts/train_diffusion.py", line 349, in main
val_loss = validate(it)
File "/home/KGDiff-main/scripts/train_diffusion.py", line 238, in validate
atom_auroc = get_auroc(np.concatenate(all_true_v), np.concatenate(all_pred_v, axis=0),
File "/home/KGDiff-main/scripts/train_diffusion.py", line 42, in get_auroc
logger.info(f'atom: {mapping[feat_mode][c]} \t auc roc: {auroc:.4f}')
NameError: name 'logger' is not defined在修改代码以后,即可正常运行训练部分的代码。再次运行
python scripts/train_diffusion.py \
--config configs/training.yml输出:
[2024-01-03 21:51:21,921::train::INFO] atom: (6, False) auc roc: 0.8272
[2024-01-03 21:51:21,924::train::INFO] atom: (6, True) auc roc: 0.9142
[2024-01-03 21:51:21,927::train::INFO] atom: (7, False) auc roc: 0.5819
[2024-01-03 21:51:21,930::train::INFO] atom: (7, True) auc roc: 0.8044
[2024-01-03 21:51:21,932::train::INFO] atom: (8, False) auc roc: 0.8133
[2024-01-03 21:51:21,935::train::INFO] atom: (8, True) auc roc: 0.6877
[2024-01-03 21:51:21,938::train::INFO] atom: (9, False) auc roc: 0.8247
[2024-01-03 21:51:21,940::train::INFO] atom: (15, False) auc roc: 0.8127
[2024-01-03 21:51:21,943::train::INFO] atom: (16, False) auc roc: 0.6230
[2024-01-03 21:51:21,946::train::INFO] atom: (16, True) auc roc: 0.7472
[2024-01-03 21:51:21,948::train::INFO] atom: (17, False) auc roc: 0.7618
[2024-01-03 21:51:21,948::train::INFO] [Validate] Iter 01000 | Loss 1.075903 | Loss pos 0.953521 | Loss v 1.011946 e-3 | Loss exp 21.187793 e-3 | Avg atom auroc 0.828030
[2024-01-03 21:51:22,058::train::INFO] [Validate] Best val loss achieved: 1.075903
[2024-01-03 21:51:42,173::train::INFO] [Train] Iter 1200 | Loss 0.973956 (pos 0.870810 | v 0.000796 | exp 0.023551) | Lr: 0.001000 | Grad Norm: 0.910338
[2024-01-03 21:52:02,200::train::INFO] [Train] Iter 1400 | Loss 0.963688 (pos 0.839717 | v 0.001212 | exp 0.002788) | Lr: 0.001000 | Grad Norm: 0.657970
[2024-01-03 21:52:22,156::train::INFO] [Train] Iter 1600 | Loss 0.445534 (pos 0.377464 | v 0.000584 | exp 0.009639) | Lr: 0.001000 | Grad Norm: 1.112845
[2024-01-03 21:52:42,524::train::INFO] [Train] Iter 1800 | Loss 1.414450 (pos 1.331138 | v 0.000740 | exp 0.009311) | Lr: 0.001000 | Grad Norm: 1.786610
[2024-01-03 21:53:02,609::train::INFO] [Train] Iter 2000 | Loss 0.257379 (pos 0.114499 | v 0.001375 | exp 0.005397) | Lr: 0.001000 | Grad Norm: 0.729617
Validate: 100%|█████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████████| 25/25 [00:06<00:00, 3.67it/s]训练过程保存在,logs_diffusion目录下以运行时间为名的文件夹内,例如:training_2024_01_03__21_49_24,文件夹内目录如下:
.
├── checkpoints
│ ├── 0.pt
│ ├── 10000.pt
│ ├── 1000.pt
│ ├── 2000.pt
│ ├── 3000.pt
│ ├── 5000.pt
│ ├── 7000.pt
│ └── 8000.pt
├── events.out.tfevents.1704336564.a01.2547340.0
├── log.txt
├── models
│ ├── common.py
│ ├── molopt_score_model.py
│ ├── __pycache__
│ │ ├── common.cpython-39.pyc
│ │ ├── molopt_score_model.cpython-39.pyc
│ │ └── uni_transformer.cpython-39.pyc
│ └── uni_transformer.py
├── training.yml
└── vis
注意:作者这一部分包含了vina?score打分等的评价,但是由于生成分子的质量较差,同时修改代码内容越来越多,比较费力,还要配置环境,我就不在这里详细描述了。这一部分的工作,可以自行使用其他工具代替。
4.6 不同引导强度下生成分子的有效性和结合力
使用4.3中生成分子的方法,按照4.4中的处理方法,测试了不同pos_grad_weight 参数下,生成分子的有效率。pos_grad_weight 参数是在分子生成过程中,坐标部分的引导强度。测试pos_grad_weight 参数的值分别为:0, 25, 50, 100,每个参数分别生成100个分子,各需要1个小时。
有效率分子统计如下:
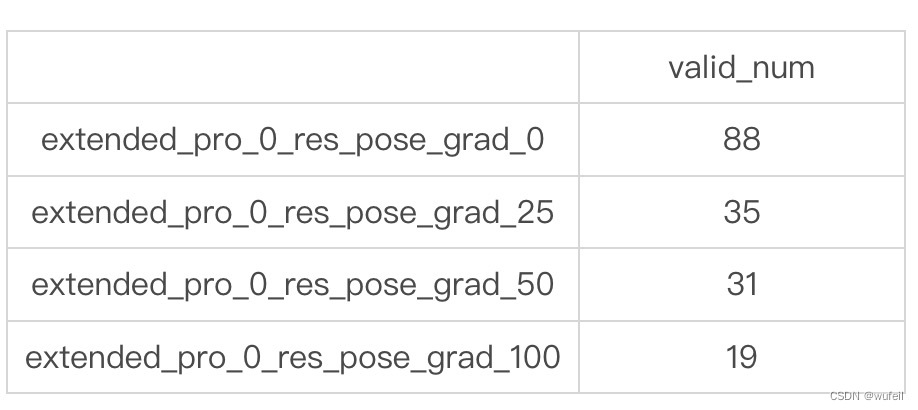
可以清晰的看到,随着坐标部分的引导强度的增加,生成分子的有效率是显著降低的。当然,关于不同引导强度的效果,作者做过测试的,所以才设定为25。
奇怪的是,在重新进行ligpr和docking以后,pos_grad_weight 参数的值为0, 25, 50, 100,最后获得的分子数分别为:8, 12, 12, 9。似乎大部分的生成的分子都无法对接。这种情况,在之前的分子生成模型中,不常见。
下图是一些对接失败分子的示例:

这些分子就算对接成功似乎也没有任何的意义,距离类药差距非常明显。
下图分别为最优打分分子的pose,及打分。
pos_grad_weight = 0, docking_score = -7.376。

pos_grad_weight = 25, docking_score = -7.157。
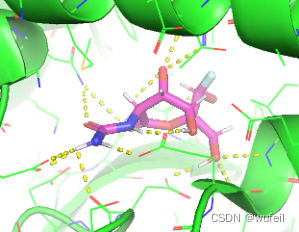
pos_grad_weight = 50, docking_score = -7.157。
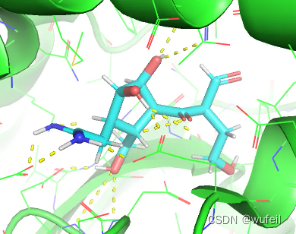
pos_grad_weight = 100, docking_score = -6.423。
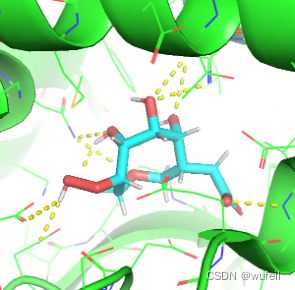
但是,晶体口袋中的分子,docking_score仅为:-4.369。
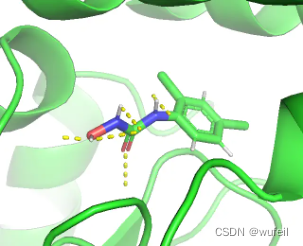
进一步查看了,其他对接成功的分子,还是有很多分子的打分优于参考分子。从这一点来说,KGDiff达到了文章中宣称的作用,Vina score 的知识引导,提升了生成分子与口袋的结合力。但是,从图中可以看到生成分子的质量,远远不能说是一个类药分子。生成的分子要比TargetDiff更差,也说明知识引导走向了极端,生成了更多的极性基团。
五、总结
KGDiff提出了一种知识引导的分子生成的扩散模型。与传统的分子生成的扩散模型不同,KGDiff的SE3等变网络的训练目标不仅是预测分子的去噪过程,而且还是预测含噪音小分子和口袋之间的结合力。在分子生成过程中,利用SE3等变网络预测的结合力,计算结合力对于原子坐标和原子类型的梯度引导分子的原子坐标和原子类型,逐步去噪,实现生成的分子与口袋之间强结合力。
测评结果显示,KGDiff在知识引导下,确实生成了结合力明显优于参考分子的分子。但是,生成分子的有效率很低,且引导强度越强,生成分子有效率越低。此外,为了增强结合力,模型不断尝试生成极性的分子,导致生成分子的类药性极差。
因此,总的来说,KGDiff是一个概念验证的文章,证明了知识引导可以生成结合力更强的分子,启发意义强,值得借鉴。
六、代码修改位置
6.1 train_diffusion.py
将get_auroc函数和get_pearsonr函数放置到main函数里面,放在main函数中trian函数之前,即157 行以后。
6.2 evaluate_diffusion.py
增加保存mol对象为sdf格式的函数,放在import 模块之后。
def save_sdf(mol, path):
writer = SDWriter(path)
writer.write(mol)
writer.close()在?mol = reconstruct.reconstruct_from_generated(pred_pos, pred_atom_type, pred_aromatic, pred_exp_atom_weight)之后添加保存mol对象为sdf文件的代码
####################################
# wufeil
# 将mol对象保存到sdf文件中
path = os.path.join(sdf_path, str(sample_idx)+'.sdf')、
save_sdf(mol, path)
###################################增加了保存smiles相关的代码,已保存生成的smiles。
smiles_list = [] # 添加的
for example_idx, r_name in enumerate(tqdm(results_fn_list, desc='Eval')):smiles = Chem.MolToSmiles(mol)
smiles_list.append(smiles) # 添加的保存smiles,成txt文件, 添加的
smile_txt_path = os.path.join(sdf_path, 'generated_molecules_smiles.txt')
with open(smile_txt_path, 'w') as file:
for line in smiles_list:
file.write(line + '\n')
logger.info(f'Evaluate done! {num_samples} samples in total.')本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- CEC2013(python):五种算法(OOA、WOA、GWO、DBO、HHO)求解CEC2013(python代码)
- 7-3 jmu-Java-05集合(泛型)-10-GeneralStack(Java for PTA)
- 人性的弱点:如何交友并影响他人
- 【面试突击】深挖网络 IO 面试实战
- 边缘计算的挑战与机遇:实时性、安全性与创新驱动
- Java中的public、private、static和final浅谈
- 基于 huggingface diffuser 库云服务器实现 stable diffusion inpaint样例代码
- 互联网行业的应届大学生,如何制作高水平简历?
- Windows Server 2003 (NT 5.2.3790.0) 构建指南
- UE5 将类修改目录