Nat. Methods | RoseTTAFoldNA准确预测蛋白质-核酸复合体
今天为大家介绍的是来自Frank DiMaio团队的一篇论文。蛋白质-核糖核酸(RNA)和蛋白质-脱氧核糖核酸(DNA)复合体在生物学中扮演着至关重要的角色。尽管近年来在蛋白质结构预测方面取得了显著进展,但预测没有同源已知复合体的蛋白质-核酸复合体的结构仍是一个基本未解决的问题。在这里,作者将RoseTTAFold机器学习蛋白结构预测方法扩展应用,以预测核酸和蛋白质-核酸复合体。作者开发了一个网络系统,RoseTTAFoldNA,它能够快速生成带有可信度估计的蛋白质-DNA和蛋白质-RNA复合体的三维结构模型。

当前预测蛋白质-核酸复合体结构的方法包括分别构建蛋白质和核酸的模型,然后使用对接计算来构建复合体。对于蛋白质部分的预测,像RoseTTAFold和AlphaFold这样的机器学习方法非常准确,而RNA结构预测则使用了蒙特卡洛抽样方法以及深度学习方法的组合。尽管在预测单个部分方面取得了进展,但蛋白质-核酸复合体结构的预测远远落后于单独预测蛋白质结构或RNA结构。作者在这份工作中着手将RoseTTAFold泛化,以模拟核酸以及蛋白质,并通过在PDB的结构上训练来学习蛋白质-核酸系统所需的许多新参数。作者新开发的RoseTTAFoldNA模型利用了与RoseTTAFold相同的数据,但额外增加了所有在PDB中的RNA、蛋白质-RNA和蛋白质-DNA复合物的数据。研究人员通过测试新近发布的核酸复合体结构,评估了RoseTTAFoldNA在预测蛋白质-核酸复合体结构方面的能力,并将其预测结果与AlphaFold和蛋白质-DNA对接计算方法的组合进行了比较。
模型结构
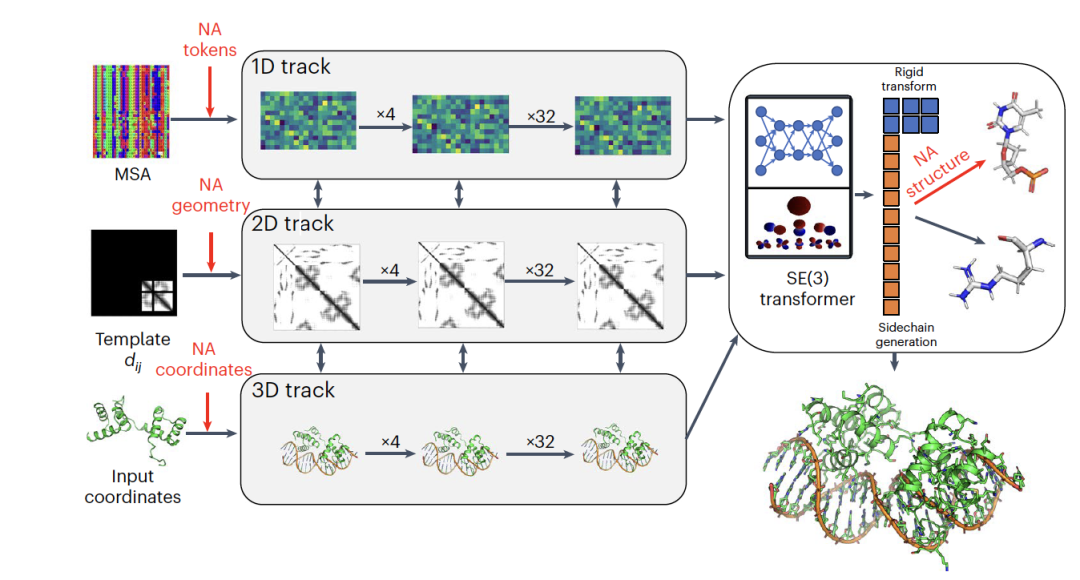
图1是RoseTTAFoldNA(RFNA)的架构,这是一种生物分子系统的三重表示方法的计算模型。RFNA基于RoseTTAFold的三轨架构,能够同时优化生物分子系统的三种表示:序列(1D,一维),残基对间距离(2D,二维)以及笛卡尔坐标(3D,三维)。RFNA对原有模型进行了几项修改以提高性能,并扩展了所有三个轨道以支持核酸和蛋白质。在RoseTTAFold的1D轨道中,原有22个标记用于蛋白质设计。RFNA在此基础上增加了10个新标记,分别对应DNA的四种核苷酸、RNA的四种核苷酸、未知DNA和未知RNA。RoseTTAFold的2D轨道能够构建蛋白质或蛋白质组合中所有氨基酸对的相互作用表示。RFNA将2D轨道泛化,以模拟核酸碱基之间以及碱基和氨基酸之间的相互作用。RoseTTAFold的3D轨道表示每个氨基酸在由三个骨架原子(N、CA和C)定义的框架中的位置和方向,并可构建多达四个侧链角。对于RFNA,还包括了对每个核苷酸的表示,使用一个坐标框架描述磷酸基团的位置和方向,并利用10个扭转角来构建核苷酸中的所有原子。RFNA包含36个这样的三轨层,后面跟着四个额外的结构细化层,总共有6700万个参数。该模型通过训练,使用蛋白质单体、蛋白质复合物、RNA单体、RNA二聚体、蛋白质-RNA复合物和蛋白质-DNA复合物,以60/40的比例处理纯蛋白质和蛋白质核酸复合的结构。
训练过程中,模型使用了基于序列相似性搜索生成的多重序列比对(MSAs),以优化网络参数。优化过程包括最小化损失函数,该函数是对所有蛋白质和核酸原子的全原子框架对齐点误差(FAPE)损失的泛化,以及评估遮蔽序列片段恢复、残基间(包括氨基酸和核苷酸)相互作用几何形状和误差预测精度的额外贡献。为了补偿PDB中含有较少核酸结构的问题,RFNA还引入了物理信息,如Lennard-Jones和氢键能量,作为最后细化层的输入特征,并在微调过程中作为损失函数的一部分。
实验结果
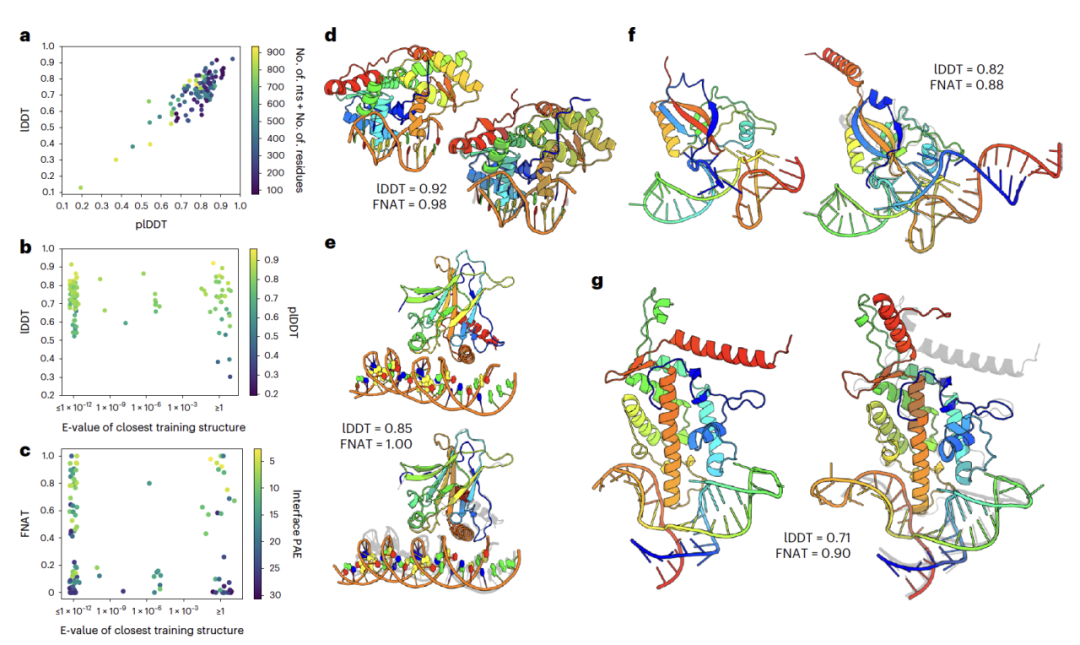
RoseTTAFoldNA在分析224个单体蛋白质-核酸(NA)复合物的表现被总结在图2中,并显示为116个簇。预测结果相当准确, Local Distance Difference Test(IDDT)得分为0.73,其中29%的lDDT得分超过0.8(19%的簇,见图2a),大约45%的结构包含超过一半的蛋白质和核酸之间的原生接触(native contacts,FNAT > 0.5,35%的簇,见图2c)。和RoseTTAFold及AlphaFold一样,RoseTTAFoldNA不仅输出预测结构,还输出模型的预测置信度。如预期,该方法能正确识别哪些结构模型是准确的。尽管只有38%的复合物(28%的簇)预测为高置信度,但在这些中,81%(78%的簇)正确地模拟了蛋白质-核酸界面(根据CAPRI标准为“可接受”或更好)。
在与训练中的蛋白质-核酸结构没有可检测序列相似性的33个簇中,准确度类似(平均lDDT = 0.68,24%的模型lDDT > 0.8,42%的FNAT > 0.5),模型仍能正确识别准确的预测——这一子集中24%的预测被预测为高置信度,其中所有8个都有根据CAPRI标准为可接受的界面。图2d-g展示了四个训练集中没有序列同源性的结构的预测,包括内切酶BpuJ1、肿瘤抗原p53、与tRNA样RNA结构域结合的SmpB,以及端粒酶逆转录酶的组分。这些预测中的不准确之处可在灵活的末端区域(图2e,g)、与界面相比DNA双螺旋的轻微倾斜(图2e)以及RNA三级结构的轻微偏差(图2f,g)中找到,但界面明显是正确的。
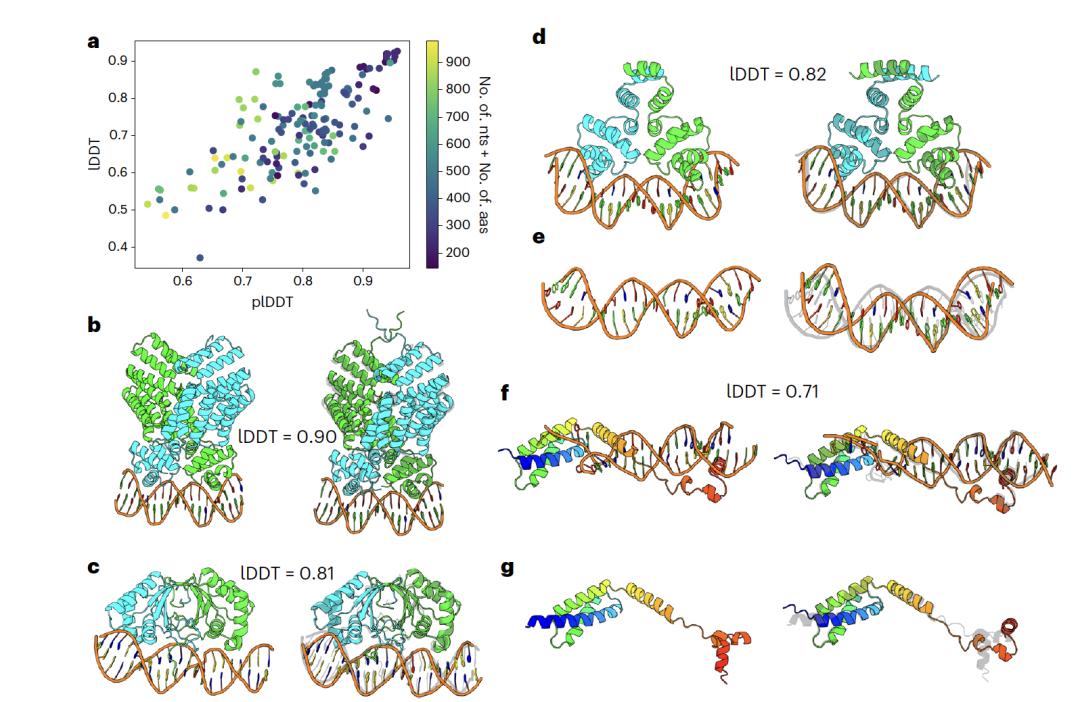
RoseTTAFoldNA的预测不仅限于只有一个蛋白质亚单位的复合物。图3总结了RoseTTAFoldNA在161个多亚单位蛋白质复合物上的性能,其中大部分是与核酸双链结合的同源二聚体蛋白。其性能与单体蛋白质-核酸复合物类似,平均lDDT = 0.72,30%的案例lDDT > 0.8,并且置信度与准确度之间有良好的一致性(图3a)。图3b-d展示了三个例子,展示了模型预测复杂结构的能力,以及蛋白质结合导致的DNA“弯曲”(图3e)。图3f,g展示了另一个例子,其中蛋白质域的相对位置只有通过共同预测这些复合物才能确定。通过首先生成独立组件的模型然后再将它们刚性地对接的方法将无法预测这种效果。
参考资料
Baek, M., McHugh, R., Anishchenko, I. et al. Accurate prediction of protein–nucleic acid complexes using RoseTTAFoldNA. Nat Methods (2023).?
https://doi.org/10.1038/s41592-023-02086-5
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- transform、 animation、 transition、 translate之间的区别
- 分布式全局ID之雪花算法
- asp.net core项目发布到 iis上
- 学生护眼台灯几瓦最好?备考好用护眼台灯推荐
- IO进程线程 day1 IO基础+标准IO
- 网站建设网络设计营销类网站eyouCMS模板(PC+WAP)
- Minio Decommission Server Pools
- 自然语言处理4——深度学习驱动情感分析 - Python高级实践
- YOLOv8全网首发:DCNv4更快收敛、更高速度、更高性能,效果秒杀DCNv3、DCNv2等 ,助力检测
- python+appium自动化测试-Appium并发测试之python启动appium服务