Sci. Rep. | 一个对任意分子体系实现准确且高效几何深度学习的通用框架
这篇工作是来自纽约城市大学/康奈尔医学院谢磊团队的一篇论文。作者提出了一个通用框架,PAMNet,可以对任意分子体系实现准确且高效的几何深度学习。在小分子性质、RNA三维结构以及蛋白质-配体结合亲和力的预测任务上,PAMNet在准确性和效率方面都优于最先进的基线模型。这些成果不仅展示了PAMNet在模拟分子体系的通用性,而且强调了其在实际分子科学应用中的潜力。

背景
分子类型和大小的多样性给药物发现、结构生物学、量子化学等领域的计算建模带来了许多挑战。为了应对这些挑战,几何深度学习(geometric deep learning, GDL)带来的最新进展变得越来越重要,尤其是图神经网络(GNN)在各种GDL方法中表现出卓越的性能。然而,现有的GNN在分子科学领域的应用仍处于初级阶段。其中一个原因是,当前的GNN通常使用有针对性的归纳偏置来建模特定类型的分子体系,从而不能被直接应用到任意其他体系,尽管所有分子结构及其相互作用都遵循相同的物理法则。此外,考虑分子中丰富几何信息的GNN往往有着高昂的计算成本,使它们难以扩展到涉及海量分子(例如高通量筛选, foundation model预训练)或大分子(例如蛋白质,RNA)的任务中。为了应对这些挑战,本文提出了一个新的通用框架:PAMNet,用于准确高效地学习不同大小和类型的三维分子的表示。PAMNet受到了分子力学的启发,明确的模拟分子中的局部和非局部相互作用及其组合效应。在涵盖不同分子体系的任务上,包括小分子性质、RNA三维结构以及蛋白质-配体结合亲和力的预测,PAMNet在准确性和效率方面都优于最先进的基线模型。近日,该项研究工作在Nature出版集团的Scientific Reports期刊上发表(DOI: 10.1038/s41598-023-46382-8),该工作的源代码已在GitHub平台上开源(https://github.com/XieResearchGroup/Physics-aware-Multiplex-GNN)。
模型概要
PAMNet的框架如图1所示。首先,基于任何分子或分子体系的三维结构,构建一个两层多重图,以分离全局和局部相互作用(图1a)。随后,PAMNet(图1b)采用多重图作为输入,并根据分子力学(图1c)的启发,通过不同的信息传递模块捕获了对应的几何信息(图1d)来模拟全局和局部相互作用。最终基于注意力机制,PAMNet整合了每种相互作用的信息,所得到的特征可用于下游任务(图1b)。此外,PAMNet在预测标量属性时利用E(3)-不变特征和操作,并可被扩展到通过考虑分子结构中的几何向量来预测E(3)-等变矢量属性。
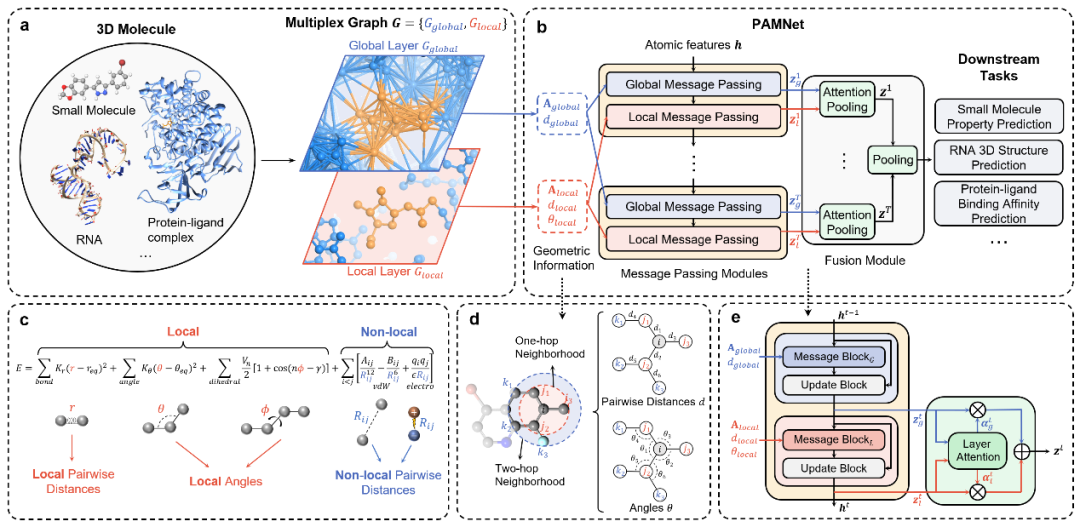
实验结果
为了全面评估PAMNet的性能,作者在不同分子体系的多种任务中进行了实验,包括小分子性质预测、RNA的三维结构预测,以及蛋白质-配体结合亲和力预测。
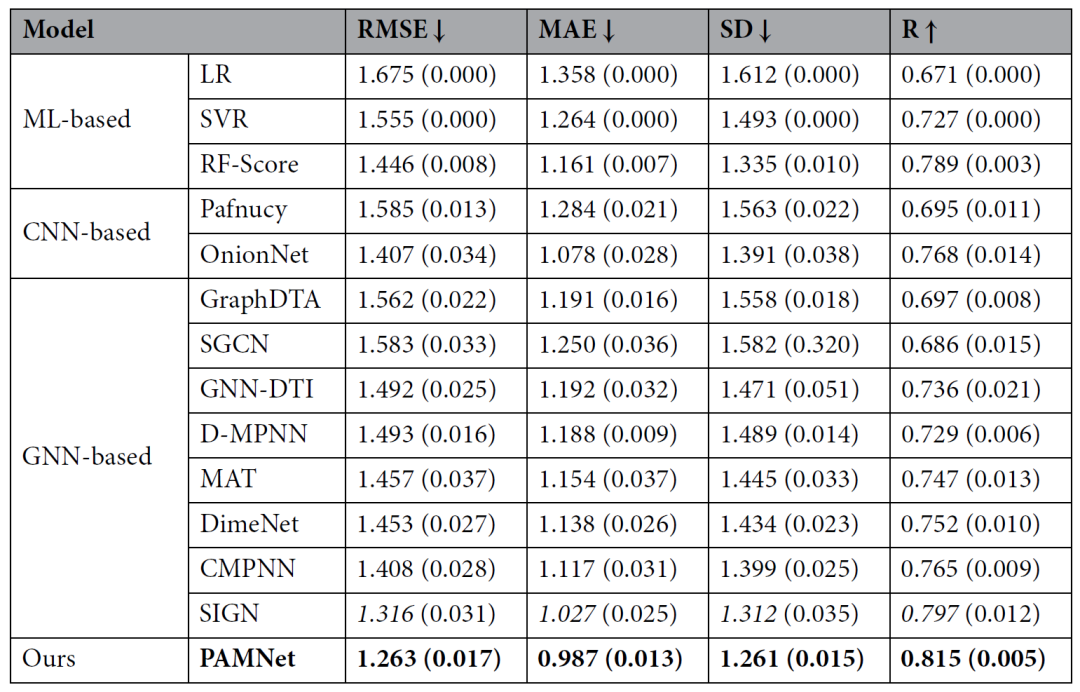
在小分子性质预测方面,PAMNet被用于预测QM9数据集中有机小分子的12种分子属性。表1的结果显示,PAMNet 在所有 12 个属性中获得了 4 个最佳结果和 6 个第二好结果。在涉及所有属性的平均绝对误差方面,PAMNet比之前的最佳模型低10%。作者还比较了一个“简单”PAMNet,称为PAMNet-s。PAMNet-s仅利用一跳邻域内的几何信息,也得到了出色的结果。
表1、QM9上的性能比较。最佳结果用粗体表示,第二好结果用斜体表示

对于RNA三维结构预测,PAMNet的任务是从一组候选的结构模型中,识别出接近真实结构的RNA模型。使用RNA-Puzzles结构预测数据集进行的测试表明,PAMNet显著优于其他四种领先的打分函数,包括ARES、Rosetta、RASP和3dRNAscore(图2)。

图2、RNA-Puzzles的性能比较。给定每个RNA的一组候选结构模型,PAMNet和其他四个打分函数对模型进行排名以进行比较。图中的每个交叉对应一个RNA。(a)比较评分函数预测的每个RNA的最佳评分结构模型。(b)比较前10佳的结构模型。(c)比较每个RNA的最佳近天然结构模型的排名。
关于蛋白质-配体结合亲和力预测,PAMNet的目标是根据每个蛋白质-配体复合物的三维结构预测其结合亲和力。在PDBbind v2016数据集上的测试中,PAMNet在多个评价指标上均优于其他基于机器学习、CNN或者GNN的方法(表2)。
表2、PDBbind上的性能比较
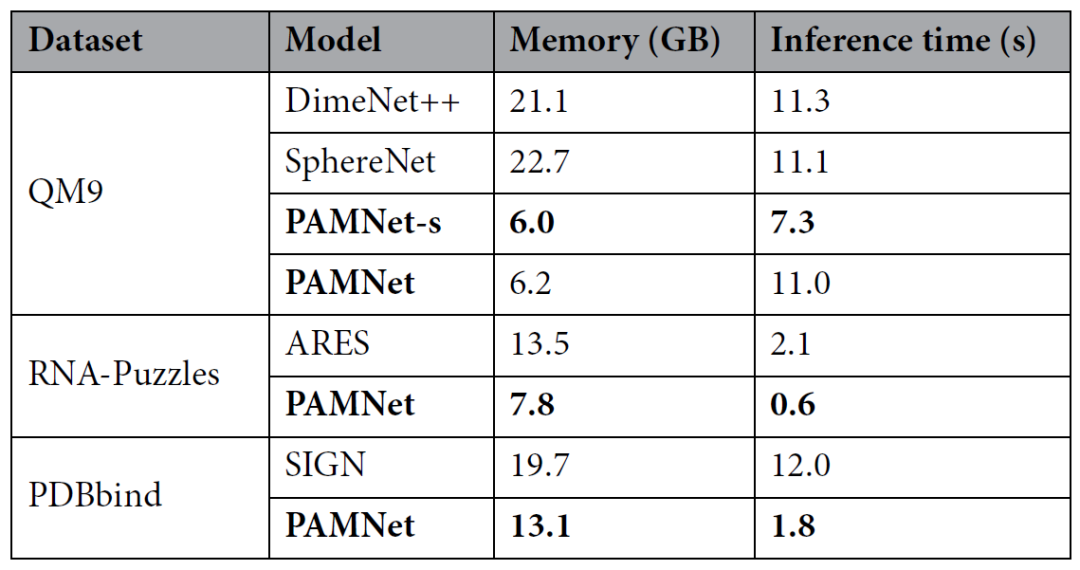
此外,为了评估PAMNet的效率,作者将其与每项任务中最准确的基准模型对于内存消耗和推理时间进行比较。结果发现,PAMNet所需的内存和推理时间都明显少于所比较的基准模型(表3)。PAMNet的高效来自于对三维分子体系中局部和非局部相互作用的单独建模:在对非局部相互作用进行建模时,作者利用了相对低复杂度的信息传递方式,仅使用原子间距作为几何信息,从而显著减少了计算复杂度。
表3、效率评估结果
结论
本研究开发了受到分子力学启发的PAMNet框架,可以对任意分子体系实现准确且高效的几何深度学习。PAMNet在多个任务中都表现出了出色的准确性和效率。这些成果不仅展示了PAMNet在模拟分子体系的通用性,而且强调了其在实际分子科学应用中的潜力。这对于未来的药物发现、结构生物学和量子化学等领域具有重要意义。
参考资料
Shuo Zhang, Yang Liu, Lei Xie. A universal framework for accurate and efficient geometric deep learning of molecular systems. Sci Rep 13, 19171 (2023).
https://doi.org/10.1038/s41598-023-46382-8
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!