用于单细胞基因调控网络分析(GRN)的SCENIC 工作流程
用于单细胞基因调控网络分析(GRN)的SCENIC 工作流程
- 摘要
- Workflow
- Pre-processing (Steps 1–4)
- Network inference (Step 5) 网络推理
- Module generation (Step 6) 模块生成
- Motif enrichment and TF-regulon prediction (cisTarget step; Step 6) 基序富集和 TF 调节子预测
- Cellular enrichment (AUCell step; Step 7) 细胞富集
- (Optional) Binarization of cellular regulon activity (可选)细胞调节子活性的二值化
- Clustering of cells based on regulon activity (Step 8) 基于调节子活性的细胞聚类
- Interactive visualization and exploration via SCope (Step 9) 通过 SCope 进行交互式可视化和探索
- Equipment setup 设备设置
- 实例
- 学习文献
摘要
这个protocol解释了如何使用软件容器和 Nextflow 管道对单细胞 RNA 测序数据执行快速 SCENIC 分析以及标准最佳实践步骤。 SCENIC 重建调节子(即转录因子及其目标基因),评估单个细胞中这些发现的调节子的活性,并使用这些细胞活动模式来寻找有意义的细胞簇。在这里,我们展示了 SCENIC 的改进版本,具有多项进步。 SCENIC 已经用 Python (pySCENIC) 进行了重构和重新实现,速度提高了十倍,并且已打包到容器中以方便使用。现在还可以使用表观基因组轨迹数据库以及基序来完善调节子。在此协议中,我们解释了 SCENIC 的不同步骤:工作流程从描述所有细胞基因丰度的计数矩阵开始,由三个阶段组成。首先,使用每目标回归方法 (GRNBoost2) 推断共表达模块。接下来,使用顺式调节基序发现 (cisTarget) 从这些模块中修剪间接目标。最后,这些调节子的活性通过调节子目标基因 (AUCell) 的富集分数进行量化。非线性投影方法可用于根据这些调节子的细胞活动模式显示细胞的视觉分组。结果可以导出为 loom 文件并在 SCope Web 应用程序中可视化。该协议通过两个用例进行说明:外周血单核细胞数据集和一组单细胞 RNA 测序癌症实验。对于包含 10,000 个基因和 50,000 个细胞的数据集,管道运行时间小于 2 小时。
重点在流程上的学习,直接到workflow部分
Workflow
简而言之,SCENIC 管道由三个步骤组成。首先,从基因之间的共表达模式推断候选调控模块(步骤 5 和 6)。接下来,通过使用 TF 基序信息消除间接目标来完善共表达模块(步骤 6)。最后,在每个单独的细胞中测量这些发现的调节子的活性并用于聚类(步骤 7 和 8;图 1)。
Pre-processing (Steps 1–4)
SCENIC 工作流程从表达矩阵开始,捕获 scRNA-seq 实验中每个细胞中每个基因转录本的丰度。通常,每个细胞在该矩阵中表示为单独的行,而基因则表示为列。虽然矩阵可以以 CSV 格式提供,但也支持用于存储单细胞基因表达谱及其元数据的digital loom file (http://loompy.org/)
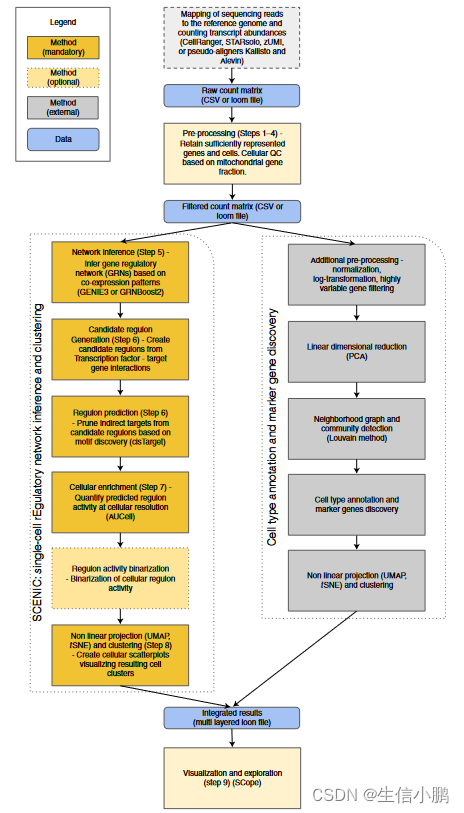
许多用于从测序读数中绘制和量化基因转录本的方法已适应单细胞方案的要求(例如 STARsolo和伪对齐器 Kallisto)。此外,还开发了 scRNA-seq 特异性方法(例如 10x Genomics 的 Cell Ranger (https://www.10xgenomics.com/)、zUMI或伪对准器 Alevin)。 SCENIC 工作流程适用于所有这些管道的输出。
在运行 SCENIC 分析之前需要进行最少量的过滤。在细胞水平上,我们检查表达基因的数量并去除处于分布极端的细胞。在外周血单核细胞 (PBMC) 研究案例中(表 1),我们丢弃表达基因 <200 个和超过 5,000 个的细胞;然而,这些阈值必须根据经验确定。我们进一步过滤掉含有大量线粒体基因转录本的细胞;这些细胞被认为质量较低,因为这表明细胞膜破裂。我们再次使用经验分布来选择线粒体基因表达的上限,这高度依赖于细胞类型,但通常为 5-15%。最后,在基因水平上,去除总体表达量低的基因;使用我们的默认设置,我们删除数据集中少于三个细胞中表达的基因。
许多工具包和指南可用于执行这些预处理步骤,其中基于 R 的 Seurat (https://satijalab.org/seurat/) 和基于 Python 的 Scanpy (https:// scanpy.readthedocs. io/en/latest/index.html)。对于此协议,我们使用 Scanpy,它高效且可扩展至大型数据集,以保持与基于 Python 的 SCENIC 版本的兼容性和互操作性。
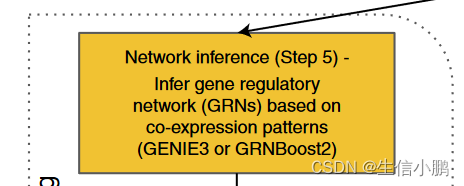
Network inference (Step 5) 网络推理
第一步,给定预定义的 TF 列表,通过基于回归的网络推理从表达或计数矩阵推断这些因素与假定目标基因之间的调控相互作用。更具体地说,对于每个表达的基因,建立一个回归模型,根据这些预定义的 TF(自变量或回归量)的表达来预测该基因在细胞中的表达(响应)。通过评估回归模型中这些因素的重要性来推断基因和 TF 之间的调控相互作用。
我们的工作流程依赖于基于树的回归模型,并且默认情况下使用基于梯度增强机回归的高效分布式实现(即 GRNBoost2)。该算法是 GENIE3 算法的重新实现,该算法基于随机森林
这些基于树的方法能够捕获组合转录控制和复杂的交互模式,总体计算负担的增加有限。在实践中,我们管道的用户仍然可以选择 GENIE3,它内置于 pySCENIC 框架中。在此步骤中可以替换其他网络推理算法,并对工作流程进行较小的调整。
此步骤的输出是将 TF 与目标基因连接起来的邻接列表。权重或重要性与这些连接相关联,以区分强监管相互作用和弱监管相互作用。
Module generation (Step 6) 模块生成
根据这些调控相互作用,根据共表达模式推断,生成模块(即 TF 及其预测的目标基因)。多种策略相结合,以便针对每个因素创建多个不同大小的模块。创建模块:
1 基于每个因素的前 N ??个目标,默认为 50 个目标。
2 基于目标的前 N ??个监管机构;默认设置选择 5、10 和 50 个调节器组(regulators)。
3 基于百分位分数;默认设置是根据因子目标的第 75 个或第 90 个百分位来选择集合。
随后,根据调节子表达与其目标之间的相关模式区分转录激活和抑制模块:正相关的目标(皮尔逊积矩相关性;默认 ρ ≥ +0.03)被分配为激活模块,而目标负相关(默认 ρ ≤ -0.03)是抑制模块的一部分。有目的地选择 0.03 的低阈值,以便 GRNBoost2/GENIE3 识别的线性以及更复杂的非线性调节相互作用不会被过滤掉并进入下一步(cisTarget - 步骤 6)。
默认情况下,在后续步骤中仅使用正相关模块,因为这些负相关模块数量较少,它们的调节相互作用权重较低,并且所得调节子总体上表现出较少的基序富集3。默认情况下(从版本 0.9.18 开始),pySCENIC 使用整个表达矩阵(包括 dropout)计算相关系数。这避免了相关系数由正在评估的两个基因中具有阳性表达的一小部分细胞确定的情况。然而,也有可能掩盖参与这两个基因中任何一个的缺失事件的细胞。
最后,所得到的具有<20个基因的模块不会被保留,因为这些模块往往不太稳定并且对目标基因丢失更敏感。我们最终得到了模块的最终集合。这些模块由调节因子 (TF) 的名称、靶基因列表(可以是直接或间接)、生成方法(顶级靶标、顶级调节因子或顶级百分位靶标)以及转录控制模式来定义(激活或抑制)。一个因子可以有多个模块,但这些模块的生成方法和/或转录模式不同。
Motif enrichment and TF-regulon prediction (cisTarget step; Step 6) 基序富集和 TF 调节子预测
模块包含调节器(regulato)的直接和间接目标,因为这些调节相互作用仅根据共表达模式推断。因此,在此步骤中,通过将模块中基因附近的顺式调控模块(CRM)分数与基因组中的其余基因进行比较,来搜索这些目标基因的假定调控区域,以寻找富集的基序。 CRM 评分是使用隐马尔可夫模型完成的,其中包含大量位置权重矩阵 (PWM),遵循 iRegulon 和 i-cisTarget 中描述的程序。如果调节因子 TF 的基序在其模块之一中显着富集,则保留该调节因子及其预测靶标以供进一步分析。
从技术上讲,cisTarget 基序富集过程依赖于排序和恢复方法(框 2):针对我们集合中与已知 TF 相关的所有基序预先计算基因组排序。三个目标物种(智人、小家鼠或果蝇)的多个版本的全基因组排名数据库可供使用 (https://resources.aertslab.org/cistarget/)。这些数据库在两个参数上略有不同:首先,基因假定调控区域的边界(例如,基因转录起始位点 (TSS) 上游 500 bp,以 TSS 为中心的 5 kb 或 10 kb),以及第二,寻找 CRM 网络级保护的直系同源物种集(例如,智人和小家鼠有 7 个或 10 个物种,黑腹果蝇有 11 个物种)。曲线下面积 (AUC) 指标用于评估给定全基因组排名的一组基因的显著恢复。
对于每个富集的基序,cisTarget 还利用此排名和恢复框架来确定恢复曲线的前沿,从而修剪模块的目标基因。最后,共享相同调节器的所有模块的直接预测靶基因组被组合成单个所得调节子。(Finally, the sets of predicted direct target genes across all modules sharing the same regulator are combined into a single resulting regulon.)
作为主题富集分析的可选补充,SCENIC 还整合了使用基于表观基因组轨迹的数据库执行富集的功能。这些是从针对 TF 的公开 ChIP-seq 实验(例如来自 ENCODE v3 的实验)生成的,生成的数据库根据与每个基因相关的调控区域的平均读取密度提供所有基因的全基因组排名。使用这些跟踪数据库,使用与基序富集相同的方法执行模块细化步骤,当上游调节器(由 GRNBoost2 预测)在其预测的目标基因上富集 ChIP-seq 峰时,选择和修剪模块。因此,通过 ChIP-seq 轨迹富集,调节子中的直接靶基因基于实际的 TF 结合位点 (TFBS)。这里使用基于轨迹的数据库是可选项,但能够提供一组独立的调节子,这些调节子可以与基于基序的调节子一起提供补充信息。
Cellular enrichment (AUCell step; Step 7) 细胞富集
类似的排序和恢复框架用于量化构成 scRNA-seq 实验的单个细胞中预测调节子的活性。具体来说,每个细胞的转录组都根据其基因的表达被建模为全基因组排名。随后通过在细胞全基因组排名上恢复其目标组来评估调节子的富集。 AUC 指标测量给定细胞中调节子的相对生物活性。
(Optional) Binarization of cellular regulon activity (可选)细胞调节子活性的二值化
调节子的细胞 AUC 值的分布有多种形状:双峰分布反映了两个不同细胞群的存在,而偏斜高斯分布代表了所检测细胞中更连续的激活模式。为了将分布的活动二值化并促进进一步的统计分析,拟合了双分量高斯混合模型来定义 AUC 阈值。对于给定的调节子,该阈值将细胞标记为“打开”或“关闭”。通过将右尾部的细胞部分标记为“on”,对偏高斯 AUC 分布进行二值化。在某些情况下,该模型无法很好地拟合特定调节子的 AUC 值分布,因此我们建议用户手动验证二值化的生物有效性。这可以通过基于网络的 SCope 可视化工具来完成(参见下面的步骤 7),该工具允许可视化分布并交互式探索阈值。
Clustering of cells based on regulon activity (Step 8) 基于调节子活性的细胞聚类
通过 AUCell 对调节子活性进行量化是一种生物降维:发现的调节子数量 (k) 通常远低于基因数量 (n),因此每个细胞可以用 k 维向量表示,而不是用 k 维向量表示由 n 维空间中的一个点。
这消除了在应用非线性投影技术(例如用于渲染视觉分组的 t 分布随机邻域嵌入 (t-SNE) 或均匀近似和投影 (UMAP) 之前)通过主成分分析 (PCA) 降低维度的需要。此外,这种基于调节子的聚类还揭示了基因表达谱背后的 GRN。
loom 文件格式是 SCENIC 管道生成的输出,是存储和交换处理的单细胞实验的通用且得到良好支持的标准。它基于数字数据存储 HDF5 (https://www.hdfgroup.org/solutions/hdf5/) 的国际标准,并提供额外的抽象层来存储表达矩阵、嵌入以及细胞和面向基因的元数据。 SCENIC 也支持 CSV,但是,对于 loom 输出格式,存储的表达矩阵通过捕获调节子(即 TF、富集基序和目标基因)的行元数据进行增强,而 AUCell 值表示为元数据与样品和色谱柱相关联。
loom 文件格式可以捕获多个嵌入,因此可用于存储其他单单元工作流程的输出。 Seurat 或 Scanpy 提供的典型工作流程是细胞类型注释和标记基因发现(框 3)。这些工作流程提供非线性细胞投影(例如,基于 PCA 的降维基因空间上的 t-SNE)和基于在细胞之间表型相似性的最近邻图抽象上运行的群落检测算法的聚类细胞。**可以根据发现的簇之间的差异表达基因来注释这些簇的细胞类型。**这些并行工作流程的所有输出都可以组合在同一个 loom 文件中,从而提供单细胞转录组学实验的全面综合概述。此工作流程的 Jupyter Notebook 和 Nextflow 实现中都提供了一个模板。
为了发现上述流程定义的细胞类型的调节因子,可以使用几种指标来衡量 SCENIC 预测的调节因子的细胞类型特异性(框 4)。
方框 4 |发现的调节子的细胞类型特异性
为了更好地表征 scRNA-seq 实验中不同细胞类型背后的调控网络,可以评估发现的调节子的细胞类型特异性。 scRNA-seq 实验中细胞的细胞类型注释可以使用框 3 中解释的一般工作流程生成,并且通常作为公开数据集的元数据提供。与细胞类型标记基因发现的一般方法类似,参数(t 检验)或非参数(Wilcoxon 秩和检验)方法可用于查找与分组的所有其他细胞类型相比,AUCell 评分具有统计显着差异的调节子一起。同样,可以依靠调节子的二值化活性来使用 Fisher 精确检验或卡方检验来查找“开启”状态与给定细胞类型同时出现的调节子。
最近,开发了一种新方法并用于量化不同细胞类型中调节子的特异性。对于这种方法,RSS 不需要对调节子的富集分数分布进行二值化,并使用 Jensen-Shannon Divergence 测量该分布与细胞类型注释的分布之间的距离。对于给定的细胞类型,所有预测调节子的 RSS 从高到低排列,高度细胞类型特异性的调节子被视为异常值。该 RSS 指标最近已合并到 SCENIC 代码库中,其使用已在链接的 Jupyter 笔记本中进行了演示。
Interactive visualization and exploration via SCope (Step 9) 通过 SCope 进行交互式可视化和探索
为了促进单细胞及其调节子的可视化和探索,该分析流程的结果可以导出并加载到 SCope 中。 SCope 是一款以交互方式可视化细胞散点图的应用程序:用户可以放大同一实验的基因、调节子和细胞类型(通过不同颜色代码标记)的活动。除了细胞的调节子(regulon)的活性,可以检查和导出调节子的富集基序和预测目标基因。此外,Scanpy 的分析结果可以与 SCENIC 结果进行比较,或者可以比较不同的 scRNA-seq 分析。它可以作为 Web 应用程序 (http://scope.aertslab.org) 提供,也可以在本地安装。
Equipment setup 设备设置
这篇文献还给了详细的安装步骤和实例,可以迁移使用。尤其是使用了doker和singularity,提高了文章的可复现性
Software installation
SCENIC 需要 Python 版本 3.6 或更高版本,并且可以使用标准 Python 包安装程序 pip (https://pip.pypa.io/en/stable/installing/) 进行安装,该安装程序从 PyPI、Python 的包索引 (https://pip.pypa.io/en/stable/installing/) 检索最新版本://pypi.org/)。为了避免包依赖性问题,建议使用隔离的 Python 环境来运行 SCENIC。这可以通过 miniconda (https://docs.conda.io/en/latest/miniconda.html) 或 pipelinev (https://github.com/pypa/pipenv) 与 virtualenv 结合来完成。
SCENIC 容器也可供下载和立即使用。在这种情况下,只要用户系统上安装了 Docker (https://www.docker.com/) 或 Singularity软件,就不需要编译或安装。可从 Docker Hub (https://hub.docker.com/) 和 Singularity Hub (https://singularity-hub.org/) 获取。
SCope 可视化工具可以按照 GitHub 存储库网站 (https://github.com/aertslab/SCope) 上提供的说明进行本地安装。
Expression matrices
表达矩阵从基因表达综合库(GEO)(https://www.ncbi.nlm.nih.gov/geo/)、ArrayExpress(https://www.ebi.ac.uk/arrayexpress/)和10x Genomics 网站 (https://support.10xgenomics.com/single-cell-gene-express/datasets)。
List of TFs
SCENIC 需要一个文本文件形式的 TF 列表,其中每行代表单个因子的基因符号。人类和小鼠 TF 列表可作为 SCENIC 的 GitHub 存储库 (https://github.com/aertslab/pySCENIC/tree/master/resources) 中的资源获取。
因子列表以智人的 HGNC 符号、小家鼠的 MGI 符号和黑腹果蝇的 FlyBase 符号的形式提供。这些因素源自 SCENIC 使用的最新版本主题集合的可用注释。对于智人,还提供了基于 Lambert 等人最近整理的限制因素列表。这些因素在网络推理步骤(步骤 5)中使用,以限制每个目标方法的基于树的回归中使用的回归器的数量。
Ranking databases
SCENIC 的 **cisTarget 步骤(步骤 6)**所依赖的排名和恢复框架需要预先计算的全基因组排名数据库作为辅助资源(框 2)。这些数据库可以从 https://resources.aertslab.org/cistarget/ 下载,适用于智人、小家鼠和黑腹果蝇。
对于每个物种,存在多个数据库,这些数据库的不同之处在于: 归因于每个基因的推定调控区域或基因组搜索空间,扫描该区域是否存在基序:基因 TSS 上游 500 bp,以基因 TSS 为中心的 5 kb TSS 或以 TSS 为中心的 10-kb。搜索空间越宽,因子靶基因的召回率越高,因为当使用更大的调节搜索空间时,也可以预测通过远距离作用增强子控制转录起始的靶标。
通过保护证实发现的基序时考虑的直系同源物种的数量。考虑的物种越多,目标的错误发现率 (FDR) 降低得越多。依靠保护来控制特异性的缺点是可能会错过物种特定的目标。这些目标基因在多样化选择下通过增强子控制,在直系同源物种中不会有相应的匹配。
用于生成数据库的主题集合的版本。该集合的最新版本 9 包含超过 24,000 个注释多个物种因子的基序,并结合了多种确定 TF 序列特异性结合模式的技术(例如文献整理、蛋白质结合微阵列或 TF 靶向 ChIP-seq)
目前有最新版本v10,并且在维护的pyscenic tutorial中建议使用V10
Motif annotations
cisTarget 步骤还需要为分析选择的排名数据库的注释表。这些表可以从 https://resources.aertslab.org/cistarget/ 下载,并将基序与结合为这些基序预测的 TFBS 的 TF 相关联。这些注释存在多个版本,应使用与用于生成数据库的版本相匹配的版本。
实例
Installation of SCENIC and configuration of environment
关键 对于每个系统安装,以下步骤只需运行一次。
- 设置Python环境。应在 Unix shell(即 bash 或类似)中运行以下示例命令来设置软件。使用Conda环境,推荐使用方式:
conda create -n scenic_protocol python=3.6
conda activate scenic_protocol
- 安装必要和可选的 Python 包并配置 Jupyter(Lab) 服务器的使用。
conda install numpy pandas matplotlib seaborn
conda install -c anaconda cytoolz
# Install scanpy (https://scanpy.readthedocs.io/en/latest/installation.html)
conda install seaborn scikit-learn statsmodels numba pytables
conda install -c conda-forge python-igraph louvain
conda install -c conda-forge multicore-tsne
pip install scanpy
# Install pySCENIC
pip install pyscenic
# Install environment as kernel for Jupyter
pip install --user ipykernel python -m ipykernel install --user --name=scenic_protocol
或者,
直接使用容器安装
如果使用容器系统,这些软件包已经完全安装在镜像中,可以通过从容器中心下载来快速获取它们。以下命令分别说明如何获取 Docker 和 Singularity 的 pySCENIC 镜像,以及对下载镜像的基本测试:
# Docker:
docker pull aertslab/pyscenic:0.9.18
# Singularity (pulls from Docker Hub):
singularity pull --name aertslab-pyscenic-0.9.18.sif \ docker://aertslab/pyscenic:0.9.18
# we recommend using a recent version of Singularity (v3 or later)
# basic usage (Singularity and Docker), interface to the command-line version of pySCENIC:
docker run -it aertslab/pyscenic pyscenic:0.9.18 -h
singularity exec aertslab-pyscenic-0.9.18.sif pyscenic -h
4 下载辅助文件:TF列表、排名数据库和主题注释(针对人类,在本例中):
# transcription factors:
wget https://raw.githubusercontent.com/aertslab/pySCENIC/master/resources/hs_hgnc_tfs.txt
# motif to TF annotation database:
wget https://resources.aertslab.org/cistarget/motif2tf/motifs-v9-nr.hgnc-m0.001-o0.0.tbl
# genome ranking database:
wget https://resources.aertslab.org/cistarget/databases/homo_sapiens/hg38/refseq_r80/mc9nr/gene_based/hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.feather
Pre-processing
- 下载案例研究的表达矩阵(来自 10x Genomics 的 PBMC 10k 数据集)。在下面的部分中,我们使用 wget Linux 实用程序从命令行获取数据集:
wget http://cf.10xgenomics.com/samples/cell-exp/3.0.0/pbmc_10k_v3/pbmc_10k_v3_filtered_fea ture_bc_matrix.tar.gz
tar xvf pbmc_10k_v3_filtered_feature_bc_matrix.tar.gz
- 对下载的表达数据集进行清洗和质量控制。我们在此示例中使用 Scanpy (https://scanpy.readthedocs.io/en/latest/) 包,并在 Python 解释器会话中运行以下语句:
import scanpy as sc
adata = sc.read_10x_mtx('filtered_feature_bc_matrix/', var_names='gene_symbols') adata.var_names_make_unique()
# compute the number of genes per cell (computes ‘n_genes' column)
sc.pp.filter_cells(adata, min_genes=0)
# mito and genes/counts cuts
mito_genes = adata.var_names.str.startswith('MT-')
# for each cell compute fraction of counts in mito genes vs. all genes
adata.obs['percent_mito'] = np.ravel(np.sum( adata[:, mito_genes].X, axis=1)) / np.ravel(np.sum(adata.X, axis=1))
# add the total counts per cell as observations-annotation to adata
adata.obs['n_counts'] = np.ravel(adata.X.sum(axis=1))
- 使用基因和细胞的基本阈值执行过滤步骤:
sc.pp.filter_cells(adata, min_genes=200) sc.pp.filter_genes(adata, min_cells=3)
adata = adata[adata.obs['n_genes'] < 4000, :]
adata = adata[adata.obs['percent_mito'] < 0.15, :]
- 使用过滤后的数据创建 loom 文件,用于下游分析
import loompy as lp
row_attrs = {
"Gene": np.array(adata.var_names),
}
col_attrs = {
"CellID": np.array(adata.obs_names),
"nGene": np.array(np.sum(adata.X.transpose()>0, axis=0)).flatten(),
"nUMI": np.array(np.sum(adata.X.transpose(),axis=0)).flatten(),
}
lp.create("PBMC10k_filtered.loom",adata.X.transpose(),row_attrs, col_attrs)
Network inference
- 从CLI 运行 GRNBoost2 网络推理算法:
在终端运行时,可能报错
FATAL: " ": executable file not found in $PATH
可能原因有可能是:
路径中的空格:有时候命令行中的额外空格会导致问题。在你的代码中,每行末尾的反斜杠 () 后面应该紧跟换行符,中间不应有空格或其他字符。
环境变量 $PATH 的问题:Singularity 在运行容器时会尝试在 $PATH 环境变量指定的目录中查找可执行文件。如果 $PATH 没有正确设置或者需要执行的命令不在 $PATH 指定的目录中,就会出现这个错误。
pyscenic grn \
--num_workers 20 \
--output adj.tsv \
--method grnboost2 \
PBMC10k_filtered.loom \
hs_hgnc_tfs.txt
使用 Docker 或 Singularity 映像时,只需在 pySCENIC 命令前添加适当的容器运行选项,包括安装适当的数据目录(此处,我们安装当前工作目录 ($PWD),假设所有数据和参考文件都位于):
## Docker
docker run -it -v $PWD:$PWD aertslab/pyscenic:0.9.18 \
pyscenic grn \
{... } # 按照上面的参数进行设置
## Singularity
singularity exec -B $PWD:$PWD aertslab-pyscenic-0.9.18.sif \
pyscenic grn \ {... } # 按照上面的参数进行设置
Candidate regulon generation and regulon prediction 候选调节子生成和调节子预测
- 从命令行执行以下语句
pyscenic ctx \
adj.tsv \
hg38__refseq-r80__10kb_up_and_down_tss.mc9nr.feather \
--annotations_fname motifs-v9-nr.hgnc-m0.001-o0.0.tbl \
--expression_mtx_fname PBMC10k_filtered.loom \
--mode "dask_multiprocessing" \
--output reg.csv \
--num_workers 20 \ # 根据硬件情况选择相应的核心数
--mask_dropouts
Cellular enrichment
- 调用以下 shell 语句来执行 AUCell 步骤:
pyscenic aucell \
PBMC10k_filtered.loom \
reg.csv \
--output PBMC10k_SCENIC.loom \
--num_workers 20
Nonlinear projection and clustering 非线性投影和聚类
- 在这里,我们说明了从调节子 AUC 矩阵生成 t-SNE 和 UMAP 降维所涉及的基本步骤。可以在关联的 Jupyter 笔记本中找到完整的工作示例
import loompy as lp
import umap from MulticoreTSNE
import MulticoreTSNE as TSNE
lf = lp.connect("PBMC10k_SCENIC.loom", mode='r+', validate=False)
auc_mtx = pd.DataFrame(lf.ca.RegulonsAUC, index=lf.ca.CellID)
lf.close()
# UMAP
runUmap = umap.UMAP(n_neighbors=10, min_dist=0.4, metric='correlation').fit_transform
dr_umap = runUmap(auc_mtx)
# tSNE
tsne = TSNE(n_jobs=20)
dr_tsne = tsne.fit_transform(auc_mtx)
Visualization in SCope
将全套分析步骤收集到 SCope 就绪的 loom 文件中是一个广泛的过程,相关 Jupyter 笔记本中已对此进行了详细记录。访问网站 (https://github.com/aertslab/SCENICprotocol) 以获取进一步说明。
总体策略是收集在该协议过程中生成的各种结果。
这包括要嵌入的降维(来自 Scanpy 和 pySCENIC)以及任何示例注释,以及 pySCENIC 调节子信息。然后将这些结果放入 loom 文件的适当属性中。这个过程可以很容易地适应包括我们的示例数据集中不存在的其他样本注释(例如,时间点、样本或基因型信息)
生成的 loom 文件可以上传到 SCope 工具 (http://scope.aertslab.org)。 SCope 的完整功能在教程 (http://scope.aertslab.org/#/Protocol_Cases/ */tutorial) 以及“预期结果”部分中进行了演示。
这篇文献还对可能出现的问题,报错进行了总结,可以翻阅。
同时,为什么采用python来进行这一系列分析,主要还是对比了相应的时间和计算速度
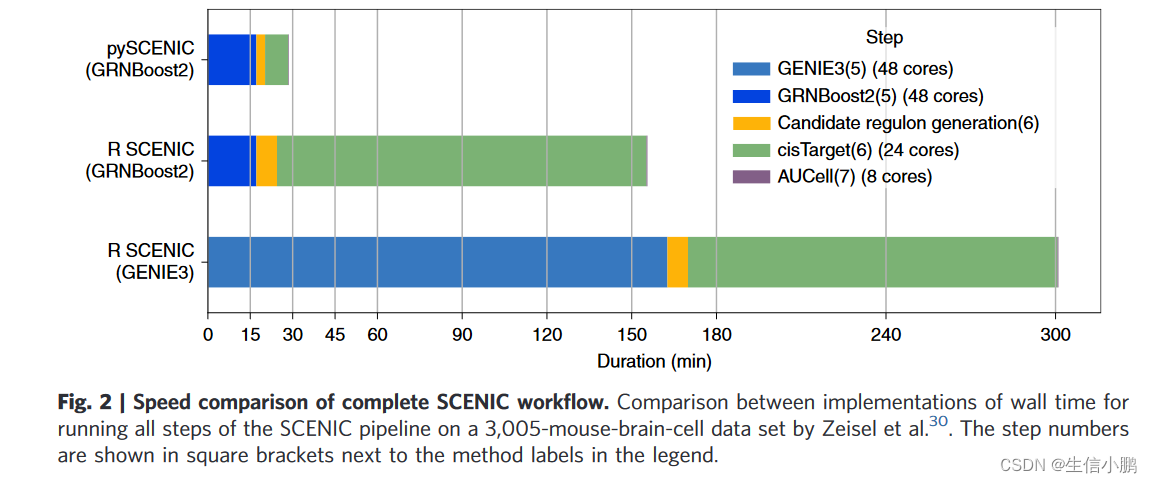
这个几乎是10倍之间的差距,还是有必要使用python进行计算学习
Anticipated results
该 SCENIC 管道的主要结果包含在一个 loom 文件中,该文件可以通过 SCope 以及以 R 或 Python 编程方式进行可视化和探索。此外,通过与 Scanpy 集成,可以在 Jupyter 笔记本中生成自定义分析和可供发布的数据。此处介绍的所有研究案例的结果均可通过以下链接在 SCope 上查看:http://scope.aertsla b.org/#/Protocol_Cases/*/welcome。 Jupyter 笔记本显示了分析这些研究案例所采取的具体完整步骤,可从 GitHub (https://github.com/aertslab/SCENICprotocol/tree/master/notebooks) 获取。
学习文献
Van de Sande, B., Flerin, C., Davie, K. et al. A scalable SCENIC workflow for single-cell gene regulatory network analysis. Nat Protoc 15, 2247–2276 (2020). https://doi.org/10.1038/s41596-020-0336-2

本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- 全球12.5m高程数据版本升级介绍
- 2. 自然数的拆分
- 适合游泳的骨传导耳机,2024年高质量游泳耳机推荐,快快入手
- 抛弃安卓,追赶iOS,鸿蒙能否成为中国第二大系统?
- 期货股市联动(期股联动助推资本市场上扬)
- Java的异常
- 计算机毕业设计----SSM自习室图书馆座位预约管理系统
- Java - 异常(三)- 声明异常(throws)和手动抛出异常throw
- HarmonyOS 应用开发学习笔记 ets组件生命周期
- Redis vs Memcached 哪一个更适合你的应用程序?