纯生信也能发7+单细胞+WGCNA+预后模型,发sci就是稳
今天给同学们分享一篇生信文章“Comprehensive analysis of scRNA-Seq and bulk RNA-Seq reveals dynamic changes in the tumor immune microenvironment of bladder cancer and establishes a prognostic model”,这篇文章发表在J Transl Med期刊上,影响因子为7.4。

结果解读:
BLCA细胞亚型的鉴定
首先,作者筛选了不合格的细胞,并得到了13,490个核心细胞用于后续分析(图1A)。对核心细胞进行基因方差分析,发现有2000个高变异基因(图1B)。对三个单细胞样本进行主成分分析(PCA)(图1C),单细胞样本呈散点分布,并得到了合理的结果。同时,在PCA中,作者还选择了20个主成分(PCs),其p值小于0.05,用于后续分析(图1D)。然后,利用umap算法将核心细胞分为19个独立的细胞亚型(图1E)。通过"singleR"软件包、CellMarker数据库和参考文献,找到标记基因对不同的细胞亚型进行注释,结果得到了七个细胞亚型,分别是B细胞、内皮细胞、T细胞、单核细胞、成纤维细胞、平滑肌细胞和上皮细胞(图1G)。通过气泡图可视化每种细胞类型的重要标记基因的表达情况(图1H)。散点图显示了不同细胞类型中标记基因的表达情况。此外,作者还研究了癌相关成纤维细胞(CAFs)中标记基因(PDPN、THY1、PDGFRB、PDGFRA和POSTN)在不同细胞类型中的表达情况,并发现所有标记基因在成纤维细胞中均高度表达。每个确定的标记基因在特定细胞中的高表达可以总结,进一步说明了细胞类型确定的可靠性。
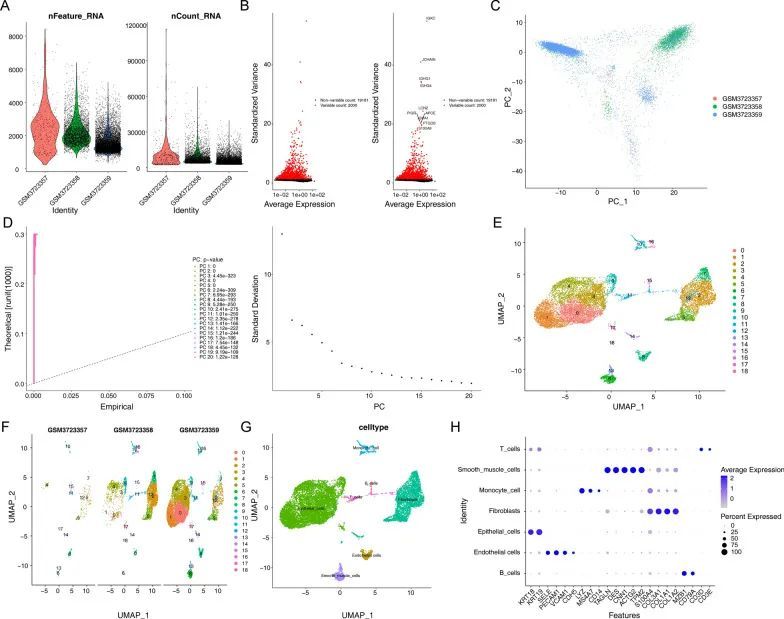
核心细胞的鉴定及其标记基因功能富集分析
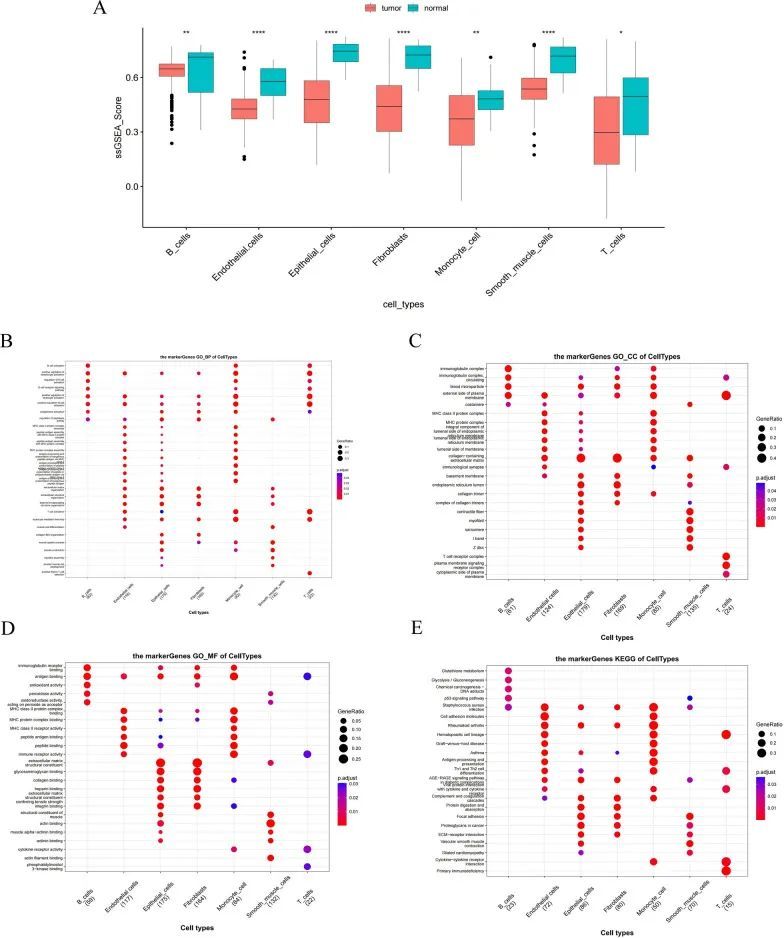
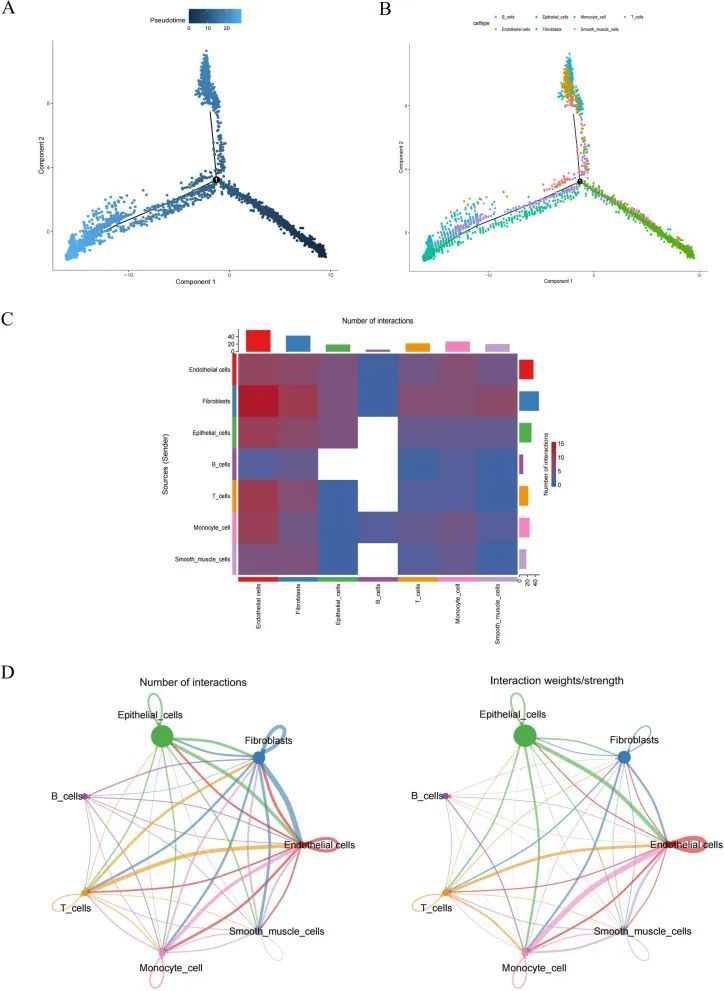
通过使用FindAllMarkers和Wilcoxon检验,获得了474个显著不同的标记基因进行鉴定。计算每个细胞显著不同的标记基因的ssGSEA得分,作者发现所有七个细胞在BLCA中都显著下调,因此这七个细胞被视为后续分析的核心细胞(图2A)。核心细胞的标记基因富集了GO和KEGG功能(图2B-E)。作者注意到,除了平滑肌细胞外,其他六种细胞类型的标记基因都与细胞激活的正调控有关,包括淋巴细胞和白细胞(图2B)。此外,单核细胞和T细胞的标记基因与细胞因子-细胞因子受体相互作用有关。B细胞的标记基因与p53信号通路相关。内皮细胞、上皮细胞和平滑肌细胞的标记基因与焦点粘附和细胞外基质受体相互作用有关(图2E)。针对所有注释的细胞,分别进行了伪时序分析,以使用Monocle 2算法探索它们的分化方向。结果显示,BLCA细胞逐渐沿着3个分化方向发展(图3A)。上皮细胞比其他细胞早期分化,并分化为两个分支,一个由内皮细胞主导,另一个由平滑肌细胞、成纤维细胞组成(图3B)。此外,作者根据特定途径和配体受体推断细胞间通讯网络以预测细胞间通讯。配体-受体对数的热图显示成纤维细胞、T细胞、单核细胞、内皮细胞和上皮细胞之间的细胞通讯更为频繁(图3C)。具体而言,内皮细胞与上皮细胞之间、内皮细胞与成纤维细胞之间、内皮细胞与T细胞之间的相互作用频率和强度较高(图3D)。此外,B细胞与其他细胞之间的相互作用相对较少。
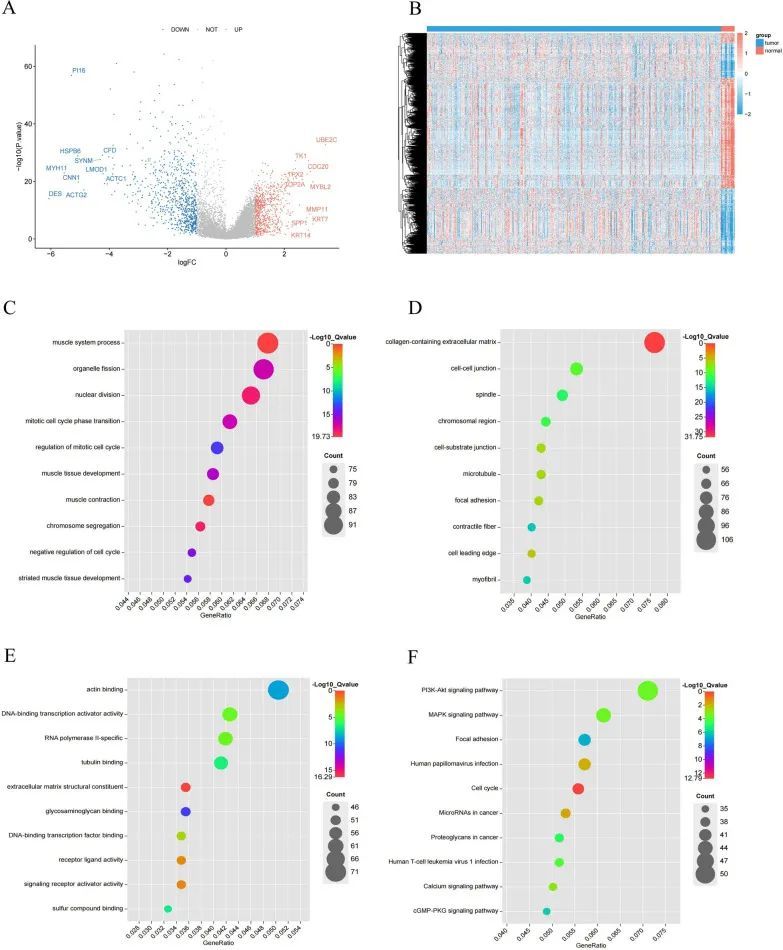
批量RNA-seq数据中差异表达基因(DEGs)的鉴定和功能富集分析
共获得1556个显著差异表达基因(DEGs),其中包括708个上调基因和848个下调基因(图4A、B)。GO分析显示,DEGs主要富集在核分裂、细胞器分裂、有丝分裂核分裂等与细胞周期相关的功能上(图4C-E)。KEGG富集结果显示,PI3K-Akt信号通路、MAPK信号通路、细胞黏附斑点和细胞周期是DEGs的富集通路(图4F)。
BLCA相关关键模块的鉴定
WGCNA被用于识别参与BLCA发展和进展的基因。在共表达网络构建过程中,当拟合指数达到0.85时,软阈值功率β设为5(图5A, A,B)。MEDissThres设为0.2,通过动态剪切树算法合并相似模块,在合并后最终得到10个模块(图5C, C,D)。根据相关系数和P值,作者选择MEbrown作为关键模块(包含2334个基因)(图5E)。关键模块基因的详细信息可见附加文件11:表S3,如图5F所示,为与临床相关性的棕色模块的散点图。
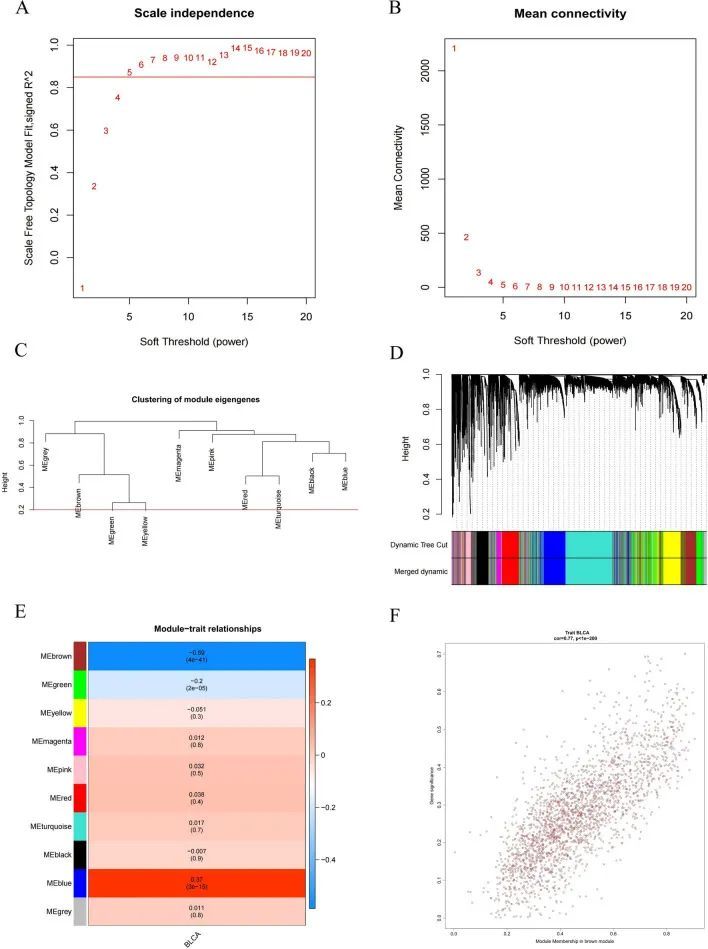
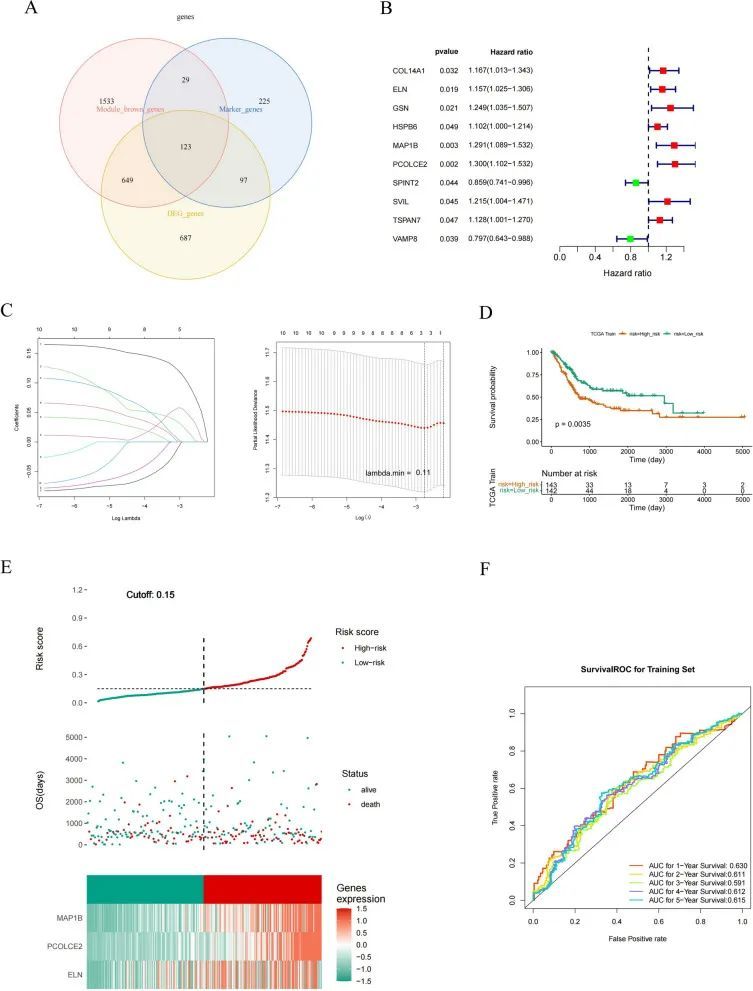
构建和验证基于3个特征基因的预后模型
使用Ven图展示了标记基因、BLCA模块基因和细胞亚型的DEGs的交集,共有123个交集基因被选取并定义为候选基因(图6A)。然后,在TCGA-BLCA的训练集中进行了单变量Cox回归分析,发现10个基因与OS显著相关(图6B)。接下来,使用LASSO算法筛选基因进行模型构建,结果如图6C所示。在最低交叉验证误差下筛选出3个特征基因:PCOLCE2、MAP1B和ELN。根据截断值=0.15,将患者分为高风险组和低风险组(图6D)。Kaplan-Meier分析显示,高风险评分患者的OS和无病生存期(DFS)显著低于低风险评分患者(图6E)。为了进一步评估风险模型的有效性,计算了OS的ROC曲线,并且1、2、3、4和5年的AUC值大于0.59,表明风险模型的疗效更好(图6F)。作者还在内部验证集和外部验证集GSE13507和GSE32548中对模型进行了功能验证,结果显示该模型具有准确性。总之,作者的预测模型在膀胱癌中显示出了出色的预测效能。
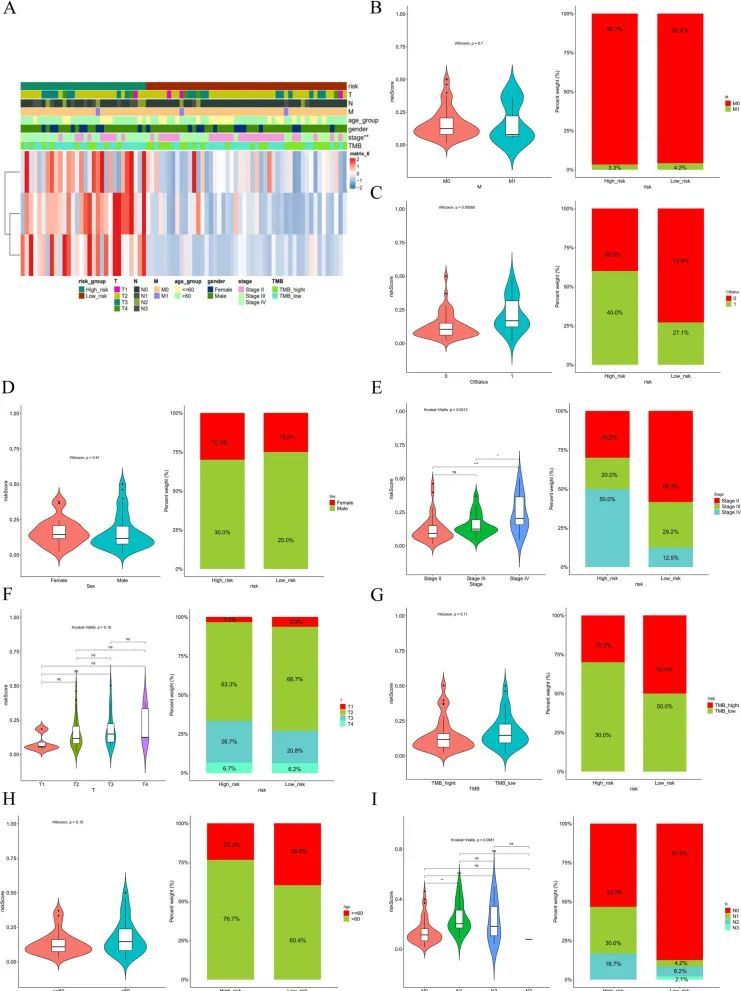
风险评分和不同临床特征的分析
为了分析风险评分表达与临床特征之间的相关性,根据不同的临床特征组别分别比较了患者的风险评分差异。结果显示,N分期、T分期和OS状态的风险评分存在显著差异(图7B)。风险模型和临床特征的热图如图7A所示。根据临床特征的分层分析显示,M0期、男性、III-IV期、T1-T2期、年龄>60岁和TMB高的患者在高风险组和低风险组的生存率上存在显著差异。综上所述,基于三个特征基因的预后模型具有良好的预后价值。
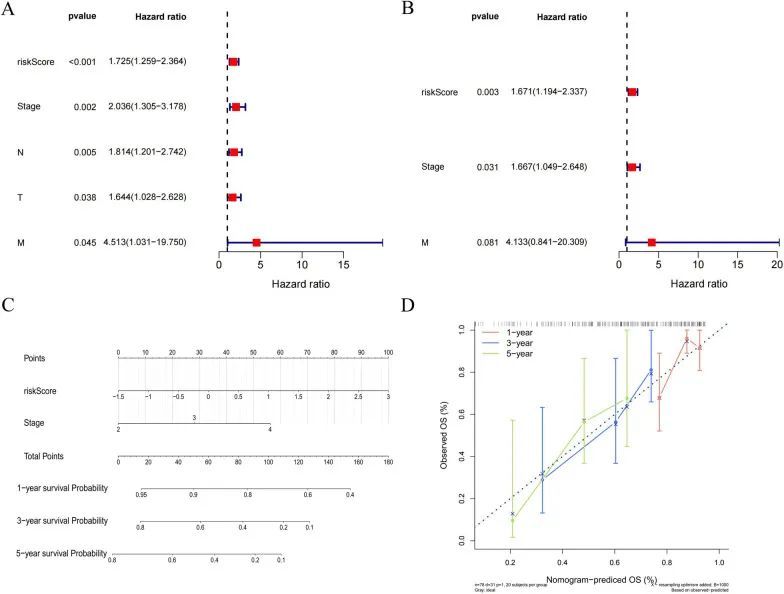
独立预后因素的筛选和绘制诊断图
为筛选独立的预后因素,临床特征和风险评分进行了单变量和多变量Cox分析。作者发现RiskScore和Stage是患者的独立预后因素(图8A,A,B)。这两个独立的预后因素被纳入了示意图模型(图8C)。此外,校准曲线显示该模型具有较高的预测效果(图8D)。因此,作者的结果表明风险评分是一个独立的预后因素,并且该示意图对预测膀胱癌患者的生存期具有较高的预测效能。
高风险组与低风险组之间的GSEA
为了分析高风险和低风险亚组对癌症进展的影响,作者进行了基因集富集分析(GSEA),以确定两组之间最显著的富集通路。结果显示,高风险组在免疫过程中显著富集,如细胞激活和参与免疫应答的体液免疫反应(图9A)。KEGG显示高风险组富集了趋化因子信号通路、补体和凝血级联反应通路,而低风险组富集了与吞噬体相关的通路(图9B)。作者还使用GSVA分析了高风险和低风险组中的所有基因。结果显示,高表达组在肌肉发生、MYC靶基因V2、早期雌激素反应、胰岛素β细胞、DNA修复、MYC靶基因V1、顶端连接、KRAS信号通路、过氧化物酶体、IL6 JAK STAT3和血管生成的MYC靶基因等标记条目中被激活,而低表达组在缺氧、脂肪生成、血红素代谢、胆汁酸代谢、干扰素α反应通路、凝血和其他标记条目中被激活(图9C、D)。
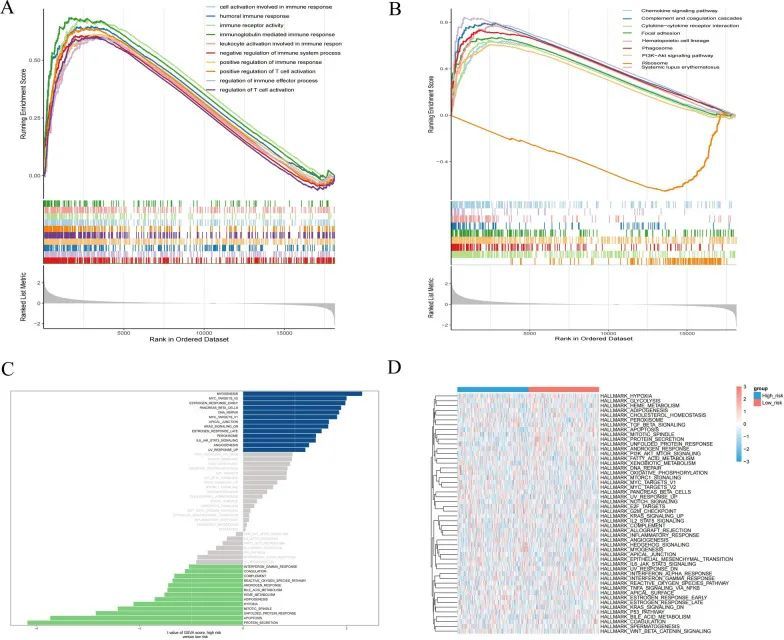
膀胱癌免疫治疗的可行性评估
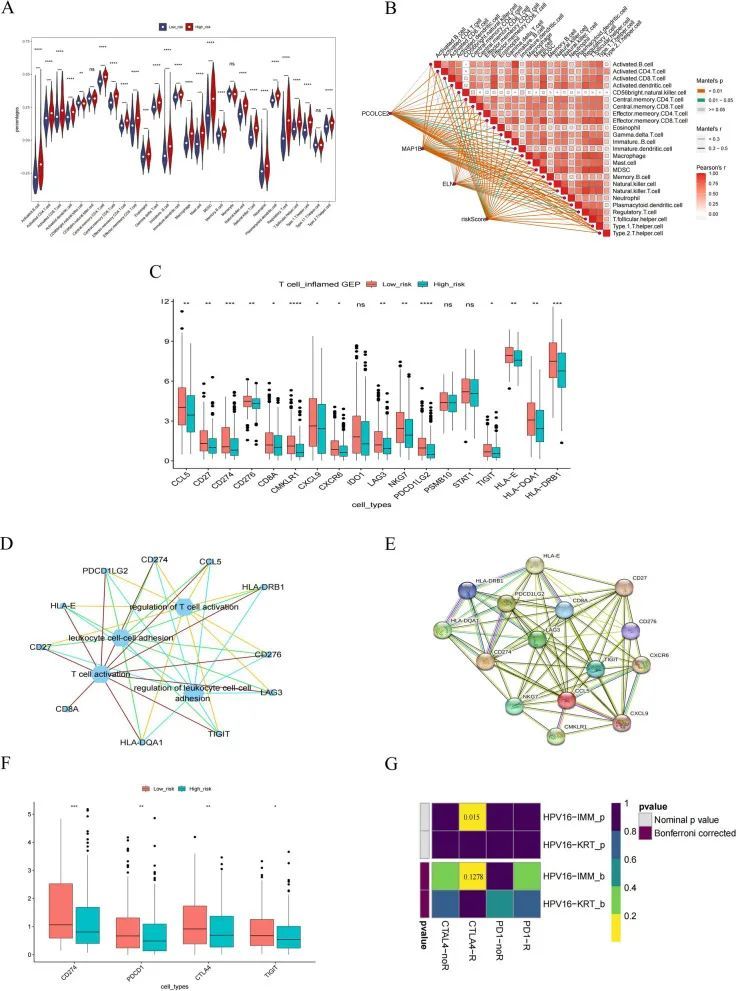
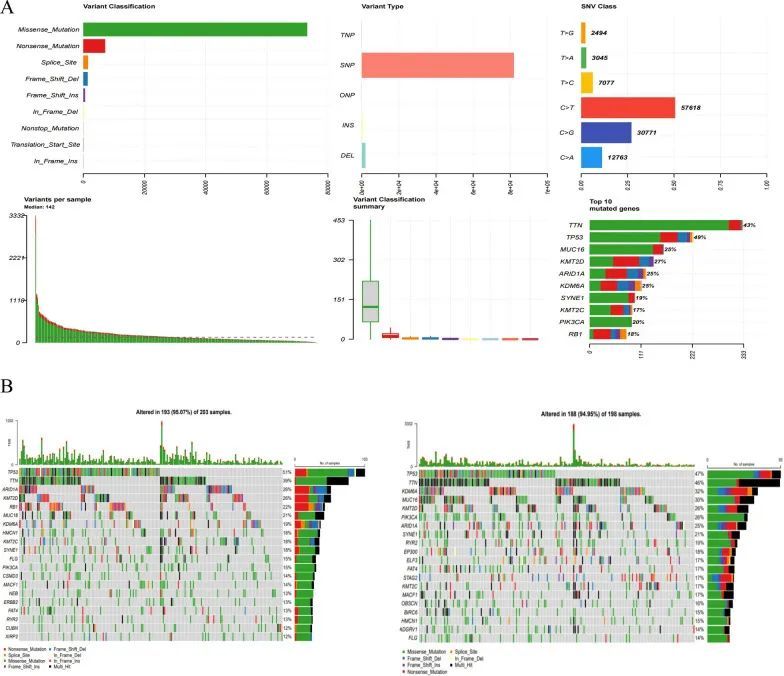
ssGSEA被用于估计不同风险组中28种免疫细胞的浸润得分。结果显示,除了自然杀伤细胞、单核细胞和T辅助细胞之外,25种免疫细胞物种的浸润水平差异在统计学上是显著的(图10A)。皮尔逊相关结果显示,预后基因和风险评分与浸润的免疫细胞显著相关(图10B)。高风险组和低风险组在16个GEP基因(炎症基因)和四个免疫检查点方面存在显著差异(图10C)。高风险组和低风险组之间的免疫细胞和16个差异性GEP基因的热图详见附加文件8:图S8。16个差异性GEP基因的相互作用网络和排名前4的通路(T细胞激活、T细胞激活调节、白细胞细胞间粘附调节、白细胞细胞间粘附)显示在图10D中,显示了GEP基因与这些通路之间的密切关联。差异性GEP基因的PPI网络显示了每个GEP基因之间的联系(图10E)。PD-1、PD-L1、CTLA-4和TIGIT在高风险组和低风险组中有所不同(图10F)。通过SubMap,在免疫治疗队列(Roh队列)中,高风险组和低风险组进行了ICB治疗,并评估了治疗反应。作者发现CTLA-4免疫位点在Roh队列中具有敏感性(图10G)。作者发现BLCA患者主要以错义突变和SNP为主(图11A)。高风险组和低风险组之间的突变结果显示,高风险组和低风险组中大多数突变类型为错义突变。高风险组中的突变比例高于低风险组,并且突变负荷指数TMB指数总体上高于低风险组(图11B)。总之,作者的结果表明,免疫治疗在BLCA中具有发展潜力。
高风险和低风险群体的药物敏感性分析
从GDSC数据库中,作者发现有12种药物与风险评分呈负相关(R < -0.4和p < 0.05),而只有前7种药物在图12A中显示。此外,作者还探索了这12种药物的靶点和途径,其中有5种药物没有相应的数据。12种化疗药物在高风险组和低风险组之间存在显著差异(图12B)。在CTRP数据库中,staurosporine、CCT036477、XL765、TGX.221和sunitinib这五种药物与风险评分呈最强的负相关(图12C)。同时,这五种药物的AUC值在高风险组和低风险组中也存在显著差异(图12D)。综上所述,这些药物可能对BLCA的治疗具有潜在的前景。

总结
通过整合单细胞RNA测序和批量RNA测序数据,作者采用了多种机器学习方法,并建立了一种新的预后模型,用于预测膀胱癌患者的生存概率。此外,风险评分是一个有前景的独立预后因子,与免疫微环境和临床病理特征密切相关。总体而言,本研究可作为膀胱癌疗效的可靠预测因子,为未来膀胱癌的靶向治疗开辟了新的途径。
本文来自互联网用户投稿,该文观点仅代表作者本人,不代表本站立场。本站仅提供信息存储空间服务,不拥有所有权,不承担相关法律责任。 如若内容造成侵权/违法违规/事实不符,请联系我的编程经验分享网邮箱:chenni525@qq.com进行投诉反馈,一经查实,立即删除!
- Python教程
- 深入理解 MySQL 中的 HAVING 关键字和聚合函数
- Qt之QChar编码(1)
- MyBatis入门基础篇
- 用Python脚本实现FFmpeg批量转换
- ? iOS技术博客:App备案指南
- 网络协议与攻击模拟_08DHCP协议
- Python 数据分析 Matplotlib篇 增加注释【plt.text() & plt.annotate()】(第3讲)
- 基于web的电影院购票系统
- [设计模式Java实现附plantuml源码~创建型] 集中式工厂的实现~简单工厂模式
- Android MVC 写法
- Jetson nano 实时性测试,使用stress-ng 和 cyclictest
- 详解MySQL中一条SQL执行过程
- C++中vector的sort用法
- python实训:简易车牌识别